Journal of Molecular Liquids ( IF 5.3 ) Pub Date : 2022-09-16 , DOI: 10.1016/j.molliq.2022.120373 Jamelah S.Al-Otaibi , Y.Sheena Mary , Y.Shyma Mary , Asmita Mondal , Nivedita Acharjee , S. Balachandar
|
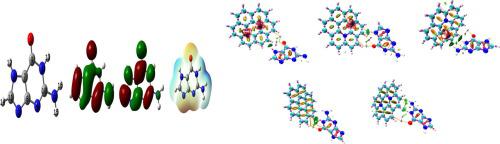
With the aid of density functional theory (DFT) and time-dependent DFT simulations, the adsorption of 2-amino-1,7-dihydropurin-6-one (ADO) on coronene and coronene that has been doped with boron and nitrogen has been investigated. While conducting this study, the binding energy, charge analysis, orbital analysis, quantum theory of atom in molecules (QTAIM) and surface enhanced Raman spectroscopy (SERS) were taken into account. The coronene sheet serves as an electron acceptor while the nucleophilic portion of ADO acts as an electron-donor, causing intermolecular interaction with a focus on the reactive region. With the exception of coronene, all the complexes have negative Gibbs free energy changes due to ADO adsorption. Furthermore, enthalpy changes have low values for coronene complex (−5.18 kcal/mol) and large values for doped complexes (−255.56 to −284. 61 kcal/mol). Our calculations demonstrate that the most stable complex among the other systems under study. Is the ADO with nitrogen parallel doped complex, with a binding energy of −284.13 kcal/mol. Other doped complexes have lower energies with the pristine coronene complex having the lowest value. The NH, NH2, C O and ring modes of ADO possess more Raman intensity in the after adsorption and it is due to the enhancement mechanism due to SERS effect.
O and ring modes of ADO possess more Raman intensity in the after adsorption and it is due to the enhancement mechanism due to SERS effect.
中文翻译:
通过 DFT 和 NCI 相互作用分析研究基于 DNA 的嘌呤衍生物在 N/B 掺杂的晕苯和晕苯上的吸附
借助密度泛函理论 (DFT) 和时间相关的 DFT 模拟,研究了 2-氨基-1,7-二氢嘌呤-6-酮 (ADO) 在掺硼和氮的晕苯和晕苯上的吸附。进行了调查。在进行这项研究时,考虑了结合能、电荷分析、轨道分析、分子中原子的量子理论 (QTAIM) 和表面增强拉曼光谱 (SERS)。晕苯片充当电子受体,而 ADO 的亲核部分充当电子供体,引起分子间相互作用,焦点集中在反应区域。除Coronene外,所有配合物由于ADO吸附而具有负吉布斯自由能变化。此外,焓变对于 Coronene 配合物具有较低的值(-5.18 kcal/mol),而对于掺杂的配合物具有较大的值(-255. 56 至 -284。61 大卡/摩尔)。我们的计算表明,在正在研究的其他系统中最稳定的复合体。是具有氮平行掺杂复合物的ADO,结合能为-284.13 kcal/mol。其他掺杂的络合物具有较低的能量,原始的coronene络合物具有最低值。新罕布什尔州,新罕布什尔州如图2所示,ADO的CO和环模式在后吸附中具有更大的拉曼强度,这是由于SERS效应的增强机制所致。






























 京公网安备 11010802027423号
京公网安备 11010802027423号