Applied Surface Science ( IF 6.3 ) Pub Date : 2022-09-14 , DOI: 10.1016/j.apsusc.2022.154910
Belinda. McFadzean , Peace Mkhonto , Phuti E. Ngoepe
|
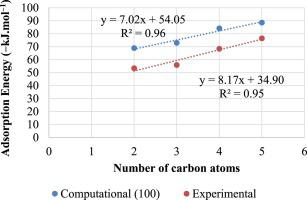
The theoretical technique of computational modelling and the experimental determination of the enthalpy of adsorption of a ligand molecule onto a mineral surface deliver the same output parameters and, therefore, complement each other in investigating the affinity of different ligand types for mineral surfaces. This study ran parallel experimental tests and computational calculations to determine the enthalpy of adsorption of a series of increasing chain length xanthates onto pyrite surfaces. We found that the vertical mode of adsorption was more exothermic than the flat mode adsorption. Most importantly we found that the flat mode adsorption for the 5C xanthate chain demonstrated the effect of van der Waals interaction of the hydrocarbons with the surface. Furthermore, the adsorption of charged xanthate both on hydrated and dry (1 0 0) surface gave inconsistent trends with experiments, except for charged xanthate flat adsorption on (1 1 1) surface. Significantly, the results of the adsorption of neutral xanthate both on dry and hydrated surfaces showed that although the absolute energy values were different; the trends were similar to the experimental approach. In particular the adsorption of neutral xanthate in water on the surface gave R2 = 0.96 and microcalorimetry gave R2 = 0.95, which indicated a linear correlation between the number of carbon atoms in the alkyl chain and the enthalpy of reaction for both computational and experimental data. The experimentally determined exothermic increase in heat of adsorption for the addition of each CH2 group was found to be −8.17 kJ.mol−1, while the contribution of the thiol head group was 34.9 kJ.mol−1. This study provides a rigorous experimental and computational approach which is intended to afford confidence in modelled data for the screening of new ligands for flotation processes.
中文翻译:
链长增加的黄原酸盐与黄铁矿表面的相互作用:DFT-D 和微量热法研究
计算建模的理论技术和配体分子在矿物表面吸附焓的实验测定提供了相同的输出参数,因此在研究不同配体类型对矿物表面的亲和力方面相互补充。这项研究进行了平行的实验测试和计算计算,以确定一系列增加链长的黄原酸盐在黄铁矿表面上的吸附焓。我们发现垂直吸附模式比平面吸附模式更放热。最重要的是,我们发现 5C 黄药链的平面模式吸附证明了烃与表面的范德华相互作用的影响。此外,带电黄药在水合和干燥(1 0 0) 表面与实验的趋势不一致,除了在 (1 1 1) 表面上的带电黄药平面吸附。值得注意的是,中性黄药在干燥和水合表面上的吸附结果表明,尽管绝对能量值不同;趋势与实验方法相似。特别是,中性黄药在水中的吸附得到 R 2 = 0.96,微量热法得到 R 2 = 0.95,这表明烷基链中的碳原子数与计算和实验的反应焓之间存在线性相关性数据。实验确定添加每种 CH 2时吸附热的放热增加发现基团为-8.17 kJ.mol -1,而硫醇头基团的贡献为34.9 kJ.mol -1。本研究提供了一种严格的实验和计算方法,旨在为筛选浮选过程的新配体的模型数据提供信心。






























 京公网安备 11010802027423号
京公网安备 11010802027423号