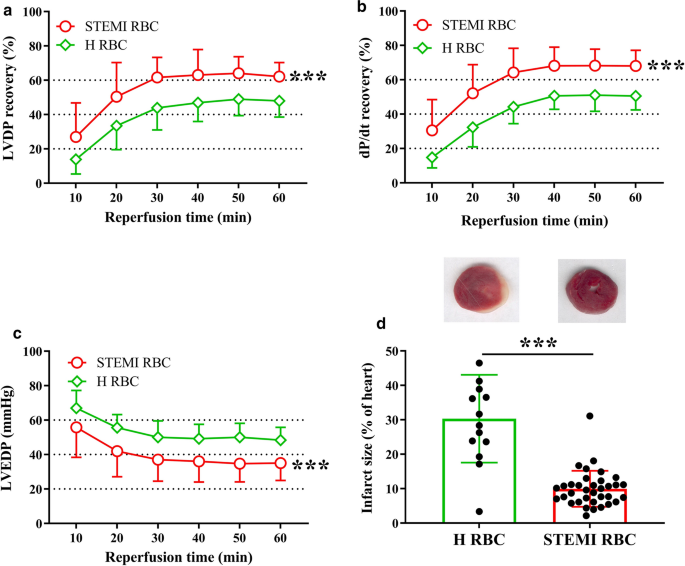
红细胞 (RBC) 被认为通过在缺氧条件下输出一氧化氮 (NO) 生物活性和 ATP 在心血管调节中发挥作用。红细胞的这种有益作用是否对急性心肌梗死患者具有保护作用尚不清楚。我们研究了来自 ST 段抬高型心肌梗死 (STEMI) 患者的红细胞是否可以防止心肌缺血再灌注损伤,以及这种作用是否涉及红细胞中的 NO 和嘌呤能信号传导。来自接受主要冠状动脉介入治疗的 STEMI 患者和健康对照者的红细胞被施用于经过全脑缺血和再灌注的离体大鼠心脏。与来自健康对照组的 RBC 相比,来自 STEMI 患者的 RBC 减少了心肌梗塞面积(30 ± 12% RBC 健康 vs. 11 ± 5% RBC STEMI 患者,P < 0.001),改善左心室发展压力和 dP/dt 的恢复,并降低经受缺血再灌注的心脏的左心室舒张末期压力。用 L-NAME 抑制 RBC NO 合酶或用 ODQ 抑制可溶性鸟苷酸环化酶 (sGC),以及抑制心肌蛋白激酶 G (PKG) 可消除心脏保护作用。此外,非选择性嘌呤能 P2 受体拮抗剂 PPADS 而不是 P1 受体拮抗剂 8PT 减弱了 STEMI 患者红细胞诱导的心脏保护作用。P2Y 13受体在红细胞中表达,P2Y 13消除了心脏保护作用受体拮抗剂 MRS2211。相比之下,在红细胞给药前灌注 PPADS、L-NAME 或 ODQ 未能阻断 STEMI 患者红细胞诱导的心脏保护作用。使用 ATP 类似物预孵育健康受试者的红细胞后,梗塞面积从 20 ± 6% 减少到 7 ± 2% ( P < 0.001),ODQ 和 MRS2211 消除了这种影响。这项研究证明了红细胞在 STEMI 患者中的一种新功能,即通过 P2Y 13受体和 NO–sGC–PKG 通路提供针对心肌缺血再灌注损伤的保护作用。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Erythrocytes from patients with ST-elevation myocardial infarction induce cardioprotection through the purinergic P2Y13 receptor and nitric oxide signaling
Red blood cells (RBCs) are suggested to play a role in cardiovascular regulation by exporting nitric oxide (NO) bioactivity and ATP under hypoxia. It remains unknown whether such beneficial effects of RBCs are protective in patients with acute myocardial infarction. We investigated whether RBCs from patients with ST-elevation myocardial infarction (STEMI) protect against myocardial ischemia–reperfusion injury and whether such effect involves NO and purinergic signaling in the RBCs. RBCs from patients with STEMI undergoing primary coronary intervention and healthy controls were administered to isolated rat hearts subjected to global ischemia and reperfusion. Compared to RBCs from healthy controls, RBCs from STEMI patients reduced myocardial infarct size (30 ± 12% RBC healthy vs. 11 ± 5% RBC STEMI patients, P < 0.001), improved recovery of left-ventricular developed pressure and dP/dt and reduced left-ventricular end-diastolic pressure in hearts subjected to ischemia–reperfusion. Inhibition of RBC NO synthase with L-NAME or soluble guanylyl cyclase (sGC) with ODQ, and inhibition of cardiac protein kinase G (PKG) abolished the cardioprotective effect. Furthermore, the non-selective purinergic P2 receptor antagonist PPADS but not the P1 receptor antagonist 8PT attenuated the cardioprotection induced by RBCs from STEMI patients. The P2Y13 receptor was expressed in RBCs and the cardioprotection was abolished by the P2Y13 receptor antagonist MRS2211. By contrast, perfusion with PPADS, L-NAME, or ODQ prior to RBCs administration failed to block the cardioprotection induced by RBCs from STEMI patients. Administration of RBCs from healthy subjects following pre-incubation with an ATP analog reduced infarct size from 20 ± 6 to 7 ± 2% (P < 0.001), and this effect was abolished by ODQ and MRS2211. This study demonstrates a novel function of RBCs in STEMI patients providing protection against myocardial ischemia–reperfusion injury through the P2Y13 receptor and the NO–sGC–PKG pathway.














































 京公网安备 11010802027423号
京公网安备 11010802027423号