Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease ( IF 4.2 ) Pub Date : 2022-08-28 , DOI: 10.1016/j.bbadis.2022.166530 Alexander Heinz 1 , Yannic Nonnenmacher 1 , Antonia Henne 1 , Michelle-Amirah Khalil 1 , Ketlin Bejkollari 1 , Catherine Dostert 2 , Shirin Hosseini 3 , Oliver Goldmann 4 , Wei He 1 , Roberta Palorini 5 , Charlène Verschueren 2 , Martin Korte 6 , Ferdinando Chiaradonna 5 , Eva Medina 4 , Dirk Brenner 7 , Karsten Hiller 1
|
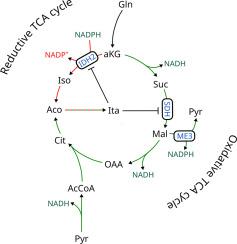
巨噬细胞在经典的促炎极化(M1 样)过程中经历广泛的代谢重编程。衣康酸盐的积累已被认为是炎症反应的结果和介质。在这项研究中,我们首先检查了分离线粒体中衣康酸盐的具体功能。我们表明 M1 巨噬细胞通过线粒体内的顺乌头酸脱羧酶 1 (ACOD1) 从头产生衣康酸。该反应的碳不仅由氧化 TCA 循环提供,而且还通过异柠檬酸脱氢酶 (IDH) 对 α-酮戊二酸的还原羧化提供。虽然巨噬细胞能够通过增强 IDH 依赖性还原羧化的活性,在缺氧期间维持一定程度的衣康酸生产,我们证明足够的衣康酸合成需要小鼠巨噬细胞中还原性和氧化性 TCA 循环代谢的平衡。相比之下,人类巨噬细胞通过增强还原羧化活性来增加低氧条件下衣康酸的积累。我们进一步证明,衣康酸可减弱 IDH2 的还原性羧化作用,从而限制其自身的产生以及免疫调节代谢物柠檬酸和 2-羟基戊二酸的积累。与此一致,在 ACOD1 耗尽的巨噬细胞中还原羧化作用增强。从机制上讲,衣康酸盐对 IDH2 的抑制与线粒体 NADP 的改变有关 人类巨噬细胞通过增强还原羧化活性来增加低氧条件下衣康酸的积累。我们进一步证明,衣康酸可减弱 IDH2 的还原性羧化作用,从而限制其自身的产生以及免疫调节代谢物柠檬酸和 2-羟基戊二酸的积累。与此一致,在 ACOD1 耗尽的巨噬细胞中还原羧化作用增强。从机制上讲,衣康酸盐对 IDH2 的抑制与线粒体 NADP 的改变有关 人类巨噬细胞通过增强还原羧化活性来增加低氧条件下衣康酸的积累。我们进一步证明,衣康酸可减弱 IDH2 的还原性羧化作用,从而限制其自身的产生以及免疫调节代谢物柠檬酸和 2-羟基戊二酸的积累。与此一致,在 ACOD1 耗尽的巨噬细胞中还原羧化作用增强。从机制上讲,衣康酸盐对 IDH2 的抑制与线粒体 NADP 的改变有关+ /NADPH 比率和竞争性琥珀酸脱氢酶抑制。综上所述,我们的研究结果扩展了促炎巨噬细胞激活期间 TCA 循环重编程的当前模型,并确定了衣康酸盐的新调节特性。
"点击查看英文标题和摘要"






























 京公网安备 11010802027423号
京公网安备 11010802027423号