当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ir and NHC Dual Chiral Synergetic Catalysis: Mechanism and Stereoselectivity in γ-Butyrolactone Formation
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2022-08-25 , DOI: 10.1021/jacs.2c07376 Bangaru Bhaskararao 1 , Madeline E Rotella 1 , Dong Yeon Kim 2 , Jung-Min Kee 3 , Kwang Soo Kim 2 , Marisa C Kozlowski 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2022-08-25 , DOI: 10.1021/jacs.2c07376 Bangaru Bhaskararao 1 , Madeline E Rotella 1 , Dong Yeon Kim 2 , Jung-Min Kee 3 , Kwang Soo Kim 2 , Marisa C Kozlowski 1
Affiliation
|
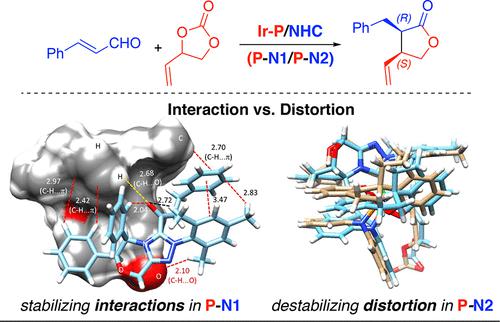
Cooperative dual catalysis is a powerful strategy for achieving unique reactivity by combining catalysts with orthogonal modes of action. This approach allows for independent control of the absolute and relative stereochemistry of the product. Despite its potential utility, the combination of N-heterocyclic carbene (NHC) organocatalysis and transition metal catalysis has remained a formidable challenge as NHCs readily coordinate metal centers. This characteristic also makes it difficult to rationalize or predict the stereochemical outcomes of these reactions. Herein, we use quantum mechanical calculations to investigate formation of γ-butyrolactones from aldehydes and allyl cyclic carbonates by means of an NHC organocatalyst and an iridium catalyst. Stereoconvergent activation of the racemic allyl cyclic carbonate forms an Ir-π-allyl intermediate and activation of an unsaturated aldehyde forms an NHC enolate, the latter of which is rate-limiting. Union of the two fragments leads to stereodetermining C–C bond formation and ultimately ring closure to generate the product lactone. Notably, CO2 loss occurs after formation of the C–C bond and Et3NH+ plays a key role in stabilizing carboxylate intermediates and in facilitating proton transfer to form the NHC enolate. The computed pathways agree with the experimental findings in terms of the absolute configuration, the enantiomer excess, and the different diastereomers seen with the (R)- and (S)-spiro-phosphoramidite combined with the NHC catalyst. Calculations reveal the lowest energy pathway includes both an NHC ligand and a phosphoramidite ligand on the iridium center. However, the stereochemical features of this Ir-bound NHC were found to not contribute to the selectivity of the process.
中文翻译:

Ir 和 NHC 双手性协同催化:γ-丁内酯形成的机理和立体选择性
协同双重催化是一种通过将催化剂与正交作用模式相结合来实现独特反应性的强大策略。这种方法允许独立控制产品的绝对和相对立体化学。尽管具有潜在的实用性,但 N-杂环卡宾 (NHC) 有机催化和过渡金属催化的结合仍然是一个艰巨的挑战,因为 N-杂环卡宾 (NHC) 很容易协调金属中心。这一特性也使得合理化或预测这些反应的立体化学结果变得困难。在此,我们使用量子力学计算来研究通过 NHC 有机催化剂和铱催化剂从醛和烯丙基环状碳酸酯形成 γ-丁内酯。外消旋烯丙基环状碳酸酯的立体会聚活化形成Ir-π-烯丙基中间体,不饱和醛的活化形成NHC烯醇化物,后者是限速的。两个片段的结合导致立体决定的 C-C 键形成,并最终闭环生成产物内酯。值得注意的是,CO 2损失发生在C-C键形成后,Et 3 NH +在稳定羧酸盐中间体和促进质子转移形成NHC烯醇化物方面发挥着关键作用。计算出的路径在绝对构型、对映体过量以及 ( R )- 和 ( S )-螺环亚磷酰胺与 NHC 催化剂结合时观察到的不同非对映体方面与实验结果一致。计算表明最低能量途径包括铱中心上的 NHC 配体和亚磷酰胺配体。 然而,这种 Ir 结合 NHC 的立体化学特征被发现对该过程的选择性没有贡献。
更新日期:2022-08-25
中文翻译:
Ir 和 NHC 双手性协同催化:γ-丁内酯形成的机理和立体选择性
协同双重催化是一种通过将催化剂与正交作用模式相结合来实现独特反应性的强大策略。这种方法允许独立控制产品的绝对和相对立体化学。尽管具有潜在的实用性,但 N-杂环卡宾 (NHC) 有机催化和过渡金属催化的结合仍然是一个艰巨的挑战,因为 N-杂环卡宾 (NHC) 很容易协调金属中心。这一特性也使得合理化或预测这些反应的立体化学结果变得困难。在此,我们使用量子力学计算来研究通过 NHC 有机催化剂和铱催化剂从醛和烯丙基环状碳酸酯形成 γ-丁内酯。外消旋烯丙基环状碳酸酯的立体会聚活化形成Ir-π-烯丙基中间体,不饱和醛的活化形成NHC烯醇化物,后者是限速的。两个片段的结合导致立体决定的 C-C 键形成,并最终闭环生成产物内酯。值得注意的是,CO 2损失发生在C-C键形成后,Et 3 NH +在稳定羧酸盐中间体和促进质子转移形成NHC烯醇化物方面发挥着关键作用。计算出的路径在绝对构型、对映体过量以及 ( R )- 和 ( S )-螺环亚磷酰胺与 NHC 催化剂结合时观察到的不同非对映体方面与实验结果一致。计算表明最低能量途径包括铱中心上的 NHC 配体和亚磷酰胺配体。 然而,这种 Ir 结合 NHC 的立体化学特征被发现对该过程的选择性没有贡献。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号