当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Chloride-Mediated Alkene Activation Drives Enantioselective Thiourea and Hydrogen Chloride Co-Catalyzed Prins Cyclizations
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2022-08-22 , DOI: 10.1021/jacs.2c06688 Dennis A Kutateladze 1 , Corin C Wagen 1 , Eric N Jacobsen 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2022-08-22 , DOI: 10.1021/jacs.2c06688 Dennis A Kutateladze 1 , Corin C Wagen 1 , Eric N Jacobsen 1
Affiliation
|
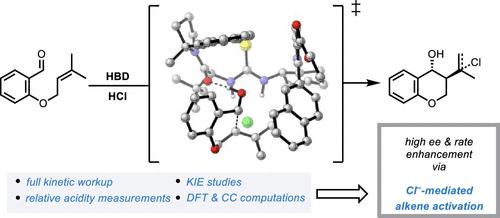
The mechanism of chiral hydrogen-bond donor (HBD) and hydrogen chloride (HCl) co-catalyzed Prins cyclizations was analyzed through a combination of experimental and computational methods and revealed to involve an unexpected and previously unrecognized mode of alkene activation. Kinetic and spectroscopic studies support the participation of a catalytically active HCl·HBD complex that displays reduced Brønsted acidity relative to HCl alone. Nevertheless, rate acceleration relative to the HCl-catalyzed background reaction as well as high levels of enantioselectivity are achieved. This inverse Brønsted correlation is ascribed to chloride-mediated substrate activation in the rate-limiting and enantiodetermining cyclization transition state. Density functional theory (DFT) calculations, distortion–interaction analysis, and quasiclassical dynamics simulations support a stepwise mechanism in which rate acceleration and enantioselectivity are achieved through the precise positioning of the chloride anion within the active site of the chiral thiourea to enhance the nucleophilicity of the alkene and provide transition-state stabilization through local electric field effects. This mode of selective catalysis through anion positioning likely has general implications for the design of enantioselective Brønsted acid-catalyzed reactions involving π-nucleophiles.
中文翻译:

氯化物介导的烯烃活化驱动对映选择性硫脲和氯化氢共催化的原环化
通过实验和计算方法相结合,分析了手性氢键供体 (HBD) 和氯化氢 (HCl) 共催化 Prins 环化的机理,并揭示了其涉及一种意想不到的且以前未被识别的烯烃活化模式。动力学和光谱研究支持催化活性 HCl·HBD 络合物的参与,该络合物相对于单独的 HCl 表现出降低的布朗斯台德酸度。尽管如此,仍实现了相对于 HCl 催化的背景反应的速率加速以及高水平的对映选择性。这种逆布朗斯台德相关性归因于限速和对映决定环化过渡态中氯介导的底物活化。密度泛函理论(DFT)计算、畸变相互作用分析和准经典动力学模拟支持逐步机制,其中通过氯阴离子在手性硫脲活性位点内的精确定位来实现速率加速和对映选择性,从而增强手性硫脲的亲核性烯烃并通过局部电场效应提供过渡态稳定。这种通过阴离子定位的选择性催化模式可能对涉及 π-亲核试剂的对映选择性布朗斯台德酸催化反应的设计具有一般意义。
更新日期:2022-08-22
中文翻译:
氯化物介导的烯烃活化驱动对映选择性硫脲和氯化氢共催化的原环化
通过实验和计算方法相结合,分析了手性氢键供体 (HBD) 和氯化氢 (HCl) 共催化 Prins 环化的机理,并揭示了其涉及一种意想不到的且以前未被识别的烯烃活化模式。动力学和光谱研究支持催化活性 HCl·HBD 络合物的参与,该络合物相对于单独的 HCl 表现出降低的布朗斯台德酸度。尽管如此,仍实现了相对于 HCl 催化的背景反应的速率加速以及高水平的对映选择性。这种逆布朗斯台德相关性归因于限速和对映决定环化过渡态中氯介导的底物活化。密度泛函理论(DFT)计算、畸变相互作用分析和准经典动力学模拟支持逐步机制,其中通过氯阴离子在手性硫脲活性位点内的精确定位来实现速率加速和对映选择性,从而增强手性硫脲的亲核性烯烃并通过局部电场效应提供过渡态稳定。这种通过阴离子定位的选择性催化模式可能对涉及 π-亲核试剂的对映选择性布朗斯台德酸催化反应的设计具有一般意义。
































 京公网安备 11010802027423号
京公网安备 11010802027423号