Journal of Physics and Chemistry of Solids ( IF 4.3 ) Pub Date : 2022-08-20 , DOI: 10.1016/j.jpcs.2022.110958 Bruno P. Silva , Antonio G.L. Costa , Mauricélio B. da Silva , Ambrósio M. Cunha , Regina C.R. Santos , Antoninho Valentini , Geancarlo Zanatta , Pedro de Lima-Neto , Ewerton W.S. Caetano , V.N. Freire
|
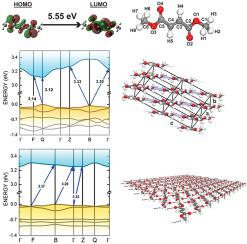
Dimethyl fumarate (DMF) is the main fumaric acid ester of the Fumaderm advanced medicament for the treatment of psoriasis and multiple sclerosis. Many properties of the DMF molecule and its crystal are still unknown. Here, the structural, electronic, and optical properties of DMF in molecular form and in the form of triclinic van der Waals crystals are addressed within the framework of density functional theory (DFT) formalism. The computations were performed using local density and generalized gradient exchange-correlation functionals, LDA and GGA, respectively, including one dispersion correction scheme for the former and two for the latter. Besides, the Δ-sol correction method was applied to improve the band-gap energy estimated from the DFT computations. The UV/Vis spectra of the DMF molecule solvated in ethanol and in the crystal were measured and compared with theoretical data obtained from time-dependent DFT (molecule) and DFT (crystal) calculations. One of the dispersion-corrected GGA functionals achieved very accurate descriptions of the lattice parameters, while analysis of the Kohn–Sham band structure indicated that the DMF crystal has a direct band-gap energy of 3.12 eV, differing from the experimental band-gap (4.00 eV) by 0.88 eV. By applying the Δ-sol correction method, the band-gap energy increased to 3.95 eV, only 0.05 eV below the experimental value. Lastly, simulations were also carried out for a single DMF monolayer, for which a band-gap of 3.26 eV was predicted. Hence, intermolecular interactions tend to decrease the electronic energy gap according to the sequence Gapcrystal < Gapmonolayer < Gapmolecule.
中文翻译:
富马酸二甲酯分子、晶体和平面:通过密度泛函理论计算的光吸收测量和结构/光电特性
富马酸二甲酯(DMF)是Fumaderm先进药物的主要富马酸酯,用于治疗银屑病和多发性硬化症。DMF 分子及其晶体的许多特性仍然未知。在这里,分子形式和三斜范德华晶体形式的 DMF 的结构、电子和光学性质在密度泛函理论 (DFT) 形式主义的框架内得到解决。计算分别使用局部密度和广义梯度交换相关泛函 LDA 和 GGA 进行,包括前者的一个色散校正方案和后者的两个色散校正方案。此外,应用 Δ-sol 校正方法来改进从 DFT 计算中估计的带隙能量。测量溶解在乙醇和晶体中的 DMF 分子的 UV/Vis 光谱,并与从时间依赖性 DFT(分子)和 DFT(晶体)计算获得的理论数据进行比较。色散校正的 GGA 泛函之一实现了对晶格参数的非常准确的描述,而对 Kohn-Sham 能带结构的分析表明,DMF 晶体的直接带隙能量为 3.12 eV,与实验带隙不同( 4.00 eV)乘以 0.88 eV。通过应用Δ-sol校正方法,带隙能量增加到3.95 eV,仅比实验值低0.05 eV。最后,还对单个 DMF 单层进行了模拟,预测的带隙为 3.26 eV。因此,分子间相互作用倾向于根据序列 Gapcrystal < 减小电子能隙




























 京公网安备 11010802027423号
京公网安备 11010802027423号