Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2022-08-13 , DOI: 10.1016/j.molstruc.2022.133928 K.M. Chandini , Fares Hezam Al-Ostoot , T.N. Lohith , Murad Q.A. Al-Gunaid , Basheer M. Al-Maswari , M.A. Sridhar , Shaukath Ara Khanum
|
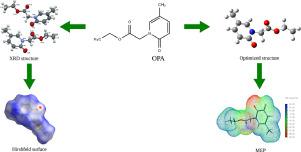
The compound ethyl 2-(5-methyl-2-oxopyridin-N-yl)acetate (OPA) has been synthesized and characterized by 1H, 13C NMR and mass spectra. The molecular structure was confirmed by single crystal X-ray diffraction studies. The compound crystallizes in triclinic crystal system with asymmetric unit Z = 2. The structure exhibits C H⋯O type of intermolecular interaction. The compound is stabilized by CH⋯π and π - π interaction. The intermolecular interactions present in the molecule are validated by Hirshfeld surface analysis and the percentage contribution from individual atoms are calculated by 2D fingerprint plots. The interaction energies are visualized through energy frameworks which show dispersion energy is predominant. The Density Functional Theory (DFT) calculations were done in gaseous, aqueous, and solvent (acetone) phase. The energy gap between the molecular orbitals HOMO and LUMO in different phases is 4.573, 4.677, 4.673 eV respectively. The charge distribution in the molecule is visualized using molecular electrostatic potential map. The noncovalent interactions present in the molecule are revealed by reduced density gradient analysis.
H⋯O type of intermolecular interaction. The compound is stabilized by CH⋯π and π - π interaction. The intermolecular interactions present in the molecule are validated by Hirshfeld surface analysis and the percentage contribution from individual atoms are calculated by 2D fingerprint plots. The interaction energies are visualized through energy frameworks which show dispersion energy is predominant. The Density Functional Theory (DFT) calculations were done in gaseous, aqueous, and solvent (acetone) phase. The energy gap between the molecular orbitals HOMO and LUMO in different phases is 4.573, 4.677, 4.673 eV respectively. The charge distribution in the molecule is visualized using molecular electrostatic potential map. The noncovalent interactions present in the molecule are revealed by reduced density gradient analysis.
中文翻译:
新型 2-(5-methyl-2-oxopyridin-N-yl) 乙酸乙酯 (OPA) 的合成、结构解析、Hirshfeld 表面分析、能量框架和 DFT 研究
合成了化合物 2-(5-甲基-2-氧代吡啶-N-基)乙酸乙酯 ( OPA),并通过1 H、13 C NMR 和质谱对其进行了表征。通过单晶 X 射线衍射研究证实了分子结构。该化合物结晶为三斜晶系,不对称单元Z =2,结构呈现C H⋯O型分子间相互作用。该化合物由 C 稳定H⋯π 和 π - π 相互作用。分子中存在的分子间相互作用通过 Hirshfeld 表面分析验证,单个原子的百分比贡献通过 2D 指纹图计算。相互作用能量通过能量框架可视化,显示分散能量占主导地位。密度泛函理论 (DFT) 计算是在气相、水相和溶剂(丙酮)相中进行的。不同相的分子轨道HOMO和LUMO之间的能隙分别为4.573、4.677、4.673 eV。使用分子静电势图可视化分子中的电荷分布。通过降低密度梯度分析揭示了分子中存在的非共价相互作用。






































 京公网安备 11010802027423号
京公网安备 11010802027423号