Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2022-08-17 , DOI: 10.1016/j.bioorg.2022.106099 Ahmed K B A W Farouk 1 , Heba Abdelrasheed Allam 1 , Essam Rashwan 2 , Riham F George 1 , Safinaz E-S Abbas 1
|
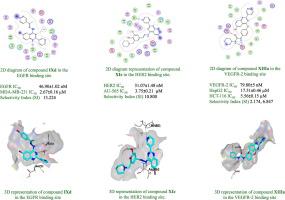
The present study involves design and synthesis of five series of 6-bromo-2-(pyridin-3-yl)-4-substituted quinazolines 9a-l, 11a-e, 13a-c, 14a-f and 15a-e. Candidates 9a-l and 11a-e were evaluated for their EGFR and HER2 inhibitory activity compared to Lapatinib. Compounds 9b, 9d, 9f, 11b and 11c were further screened for their in vitro cytotoxicity against two human breast cancer cell lines: AU-565 and MDA-MB-231 in addition to normal breast cell line MCF10A. Compound 9d revealed a remarkable cytotoxic efficacy against AU-565 cell line (IC50 = 1.54 µM) relative to Lapatinib (IC50 = 0.48 µM), whereas compounds 9d and 11c showed a superior cytotoxicity towards MDA-MB-231 (IC50 = 2.67 and 1.75 µM, respectively) in comparison to Lapatinib (IC50 = 9.29 µM). Moreover, compounds 13a-c, 13a-c, 14a-f and 15a-e were tested for their VEGFR-2 inhibitory activity compared to Sorafenib. Compounds 13a, 14c and 14e exhibited remarkable inhibition (IC50 = 79.80, 50.22 and 78.02 nM, respectively) relative to Sorafenib (IC50 = 51.87 nM). In vitro cytotoxicity of these compounds against HepG2, HCT-116 and normal cell (WISH) revealed a superior cytotoxicity against HepG2, HCT-116 especially 13a (IC50 = 17.51 and 5.56 µM, respectively) and 14c (IC50 = 10.40 and 3.37 µM, respectively) compared to Sorafenib (IC50 = 19.33 and 6.82 µM, respectively). Compounds 9d, 11c and 14c were subjected to cell cycle analysis and apoptotic assay. Molecular docking and ADME prediction studies were fulfilled to illustrate the interaction of the potent derivatives with the hot spots of the active site of EGFR, HER2 and VEGFR-2 along with prediction of their pharmacokinetic and physicochemical properties.
中文翻译:
6-bromo-2-(pyridin-3-yl)-4-取代喹唑啉作为多酪氨酸激酶抑制剂的设计与合成
本研究涉及设计和合成五个系列的 6-bromo-2-(pyridin-3-yl)-4-取代的喹唑啉9a-l、11a-e、13a-c、14a-f和15a-e。与拉帕替尼相比,评估了候选者9a-l和11a-e的 EGFR 和 HER2 抑制活性。除了正常乳腺细胞系MCF10A之外,化合物9b、9d、9f、11b和11c针对两种人乳腺癌细胞系:AU-565和MDA-MB-231进一步筛选了它们的体外细胞毒性。化合物9d显示相对于拉帕替尼(IC 50 = 0.48 µM)对 AU-565 细胞系 (IC 50 = 1.54 µM)具有显着的细胞毒性功效 ,而化合物9d和11c对 MDA-MB-231 显示出优异的细胞毒性 (IC 50 = 2.67和 1.75 µM)与拉帕替尼(IC 50 = 9.29 µM)相比。此外,与索拉非尼相比,测试了化合物13a-c、13a-c、14a-f和15a-e的 VEGFR-2 抑制活性。化合物13a、14c和相对于索拉非尼(IC 50 = 51.87 nM) , 14e表现出显着的抑制作用(IC 50 = 79.80、50.22 和 78.02 nM )。这些化合物对 HepG2、HCT-116 和正常细胞 (WISH) 的体外细胞毒性显示对 HepG2、HCT-116 尤其是13a(IC 50 = 17.51 和 5.56 µM)和14c(IC 50 = 10.40 和 3.37 )具有优异的细胞毒性µM,分别)与索拉非尼相比(IC 50 = 19.33 和 6.82 µM,分别)。化合物9d、11c和14c进行细胞周期分析和凋亡测定。通过分子对接和 ADME 预测研究来说明强效衍生物与 EGFR、HER2 和 VEGFR-2 活性位点热点的相互作用以及对其药代动力学和物理化学性质的预测。





























 京公网安备 11010802027423号
京公网安备 11010802027423号