Journal of Solid State Chemistry ( IF 3.2 ) Pub Date : 2022-08-04 , DOI: 10.1016/j.jssc.2022.123449 Xiaoxiao Zhang , Pingjian Wang , Qingyu Wu , Ling Xu , Mingyu Chen , Yunxin Kang , Chengshuai Sun , Guangfen Wei , Zhuhui Qiao , Zhonghai Lin
|
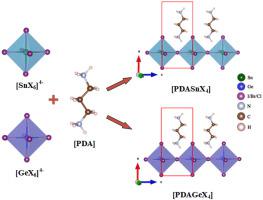
Hybrid lead (Pb) halide perovskites are widely regarded as promising materials for next generation of photovoltaic (PV) and optoelectronic (OE) devices owing to their high power conversion efficiency (PCE). To obtain green lead-free and excellent materials, the structural, electronic and optical properties of tin (Sn) and germanium (Ge) based single-layer Dion-Jacobson (DJ) phase perovskites (PDA)MX4 (M = Sn/Ge; X = I/Br/Cl) systems are investigated by carrying out first-principles theory. The calculation results indicate that all perovskites (PDA)MX4 have good thermal stability (at 300K), and the structural stability increases in the order of I–Br–Cl. These compounds are direct bandgap semiconductors, ranging from 1.26 to 1.81 eV ((PDA)SnX4) and 1.44–2.26 eV ((PDA)GeX4). Density of states (DOS) reveals that the band edge state mainly comes from the M-X bond and the organic molecule PDA is not directly involved in the contribution. Compared to Ge-based perovskites, the bandgap range of Sn-based perovskites is not only closer to the optimal bandgap requirement for perovskite solar cells (PSCs), but the optical properties reveal a slightly higher absorption coefficient for Sn-based perovskites in the visible light range. This suggests that (PDA)SnX4 perovskites are more favorable for PSCs applications. The theoretical study can provide strong support for the development of high-level perovskites materials in PV and OE devices.
中文翻译:
Dion-Jacobson 相无铅卤化物 (PDA)MX4 (M=Sn/Ge; X=I/Br/Cl) 钙钛矿:第一性原理理论
混合铅(Pb)卤化物钙钛矿因其高功率转换效率(PCE)而被广泛认为是下一代光伏(PV)和光电(OE)器件的有前途的材料。为获得绿色无铅优良材料,锡(Sn)和锗(Ge)基单层Dion-Jacobson(DJ)相钙钛矿(PDA)MX 4(M = Sn )的结构、电子和光学性能/Ge; X = I/Br/Cl) 系统通过执行第一性原理理论进行研究。计算结果表明,所有钙钛矿(PDA)MX 4均具有良好的热稳定性(300K),且结构稳定性以I-Br-Cl的顺序增加。这些化合物是直接带隙半导体,范围从 1.26 到 1.81 eV ((PDA)SnX 4) 和 1.44–2.26 eV ((PDA)GeX 4 )。态密度(DOS)表明带边缘态主要来自MX键,有机分子PDA不直接参与贡献。与 Ge 基钙钛矿相比,Sn 基钙钛矿的带隙范围不仅更接近钙钛矿太阳能电池 (PSC) 的最佳带隙要求,而且其光学特性表明 Sn 基钙钛矿在可见光波段的吸收系数略高。光范围。这表明 (PDA)SnX 4钙钛矿更适合 PSC 应用。该理论研究可为开发用于光伏和光电器件的高级钙钛矿材料提供有力支持。






























 京公网安备 11010802027423号
京公网安备 11010802027423号