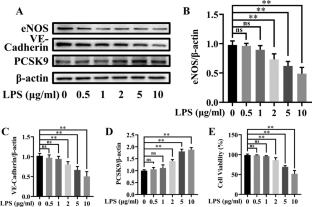
内皮功能障碍通常伴随脓毒症。我们旨在探讨 PCSK9 在脓毒性内皮功能障碍中的作用。脓毒症是由脂多糖 (LPS) 处理体外人脐静脉内皮细胞 (HUVEC)和小鼠体内盲肠结扎穿孔 (CLP) 手术引起的。Evolocumab (EVC) 和 Pep 2–8,PCSK9 抑制剂,随后被用于确定 PCSK9 在体外和体内脓毒症诱导的内皮功能障碍中的作用, 分别。此外,TLR4 激动剂 Kdo2-Lipid A 铵 (KLA) 用于确定相关机制。通过蛋白质印迹法检测小鼠主动脉和 HUVEC 中 eNOS、VE-cadherin、PCSK9、TLR4、MyD88、p-p65、p65、NLRP3、ASC 和 caspase-1 p20 的蛋白表达,同时检测 TNFα、IL-1β 的 mRNA 表达, 和 IL-18 由 qRT-PCR 确定。小鼠主动脉炎性细胞因子的水平通过免疫组织化学可视化。通过血管反应性实验检测主动脉的血管舒张。评估 CLP 后的 96 小时存活率。我们的研究结果表明,在脓毒症 HUVEC 或小鼠中,eNOS 和 VE-钙粘蛋白的表达减少,PCSK9 表达增加。抑制 PCSK9 会增加 eNOS 和 VE-钙粘蛋白的表达。TLR4/MyD88/NF-κB 和 NLRP3 通路的激活可能是脓毒症中 PCSK9 诱导的内皮功能障碍的原因。血管反应性试验和生存研究表明,抑制 PCSK9 可以防止内皮依赖性血管舒张功能下降,提高脓毒症小鼠的生存率。总之,我们的结果表明,脓毒症期间 PCSK9 表达增加会激活 TLR4/MyD88/NF-κB 和 NLRP3 通路以诱导炎症,从而导致血管内皮功能障碍并降低存活率。因此,抑制 PCSK9 可能是改善脓毒症血管内皮功能的潜在临床治疗靶点。血管反应性试验和生存研究表明,抑制 PCSK9 可以防止内皮依赖性血管舒张功能下降,提高脓毒症小鼠的生存率。总之,我们的结果表明,脓毒症期间 PCSK9 表达增加会激活 TLR4/MyD88/NF-κB 和 NLRP3 通路以诱导炎症,从而导致血管内皮功能障碍并降低存活率。因此,抑制 PCSK9 可能是改善脓毒症血管内皮功能的潜在临床治疗靶点。血管反应性试验和生存研究表明,抑制 PCSK9 可以防止内皮依赖性血管舒张功能下降,提高脓毒症小鼠的生存率。总之,我们的结果表明,脓毒症期间 PCSK9 表达增加会激活 TLR4/MyD88/NF-κB 和 NLRP3 通路以诱导炎症,从而导致血管内皮功能障碍并降低存活率。因此,抑制 PCSK9 可能是改善脓毒症血管内皮功能的潜在临床治疗靶点。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
PCSK9 Promotes Endothelial Dysfunction During Sepsis Via the TLR4/MyD88/NF-κB and NLRP3 Pathways
Endothelial dysfunction often accompanies sepsis. We aimed to explore the role of PCSK9 in septic endothelial dysfunction. Sepsis was induced by lipopolysaccharide (LPS) treatment of human umbilical vein endothelial cells (HUVECs) in vitro and cecal ligation and puncture (CLP) surgery in mice in vivo. Evolocumab (EVC) and Pep 2–8, PCSK9 inhibitors, were subsequently used to determine the role of PCSK9 in sepsis-induced endothelial dysfunction in vitro and in vivo, respectively. In addition, the TLR4 agonist, Kdo2-Lipid A ammonium (KLA), was used to determine the related mechanism. Protein expression of eNOS, VE-cadherin, PCSK9, TLR4, MyD88, p-p65, p65, NLRP3, ASC, and caspase-1 p20 in mice aortas and HUVECs was measured by western blotting, while mRNA expression of TNFα, IL-1β, and IL-18 was determined by qRT-PCR. The level of inflammatory cytokines of mouse aortas was visualized by immunohistochemistry. Vasodilation of the aorta was detected by vascular reactivity experiments. The 96-h survival rate after CLP was assessed. Our findings showed that the expression of eNOS and VE-cadherin decreased, and PCSK9 expression increased, in septic HUVECs or mice. Inhibition of PCSK9 increased eNOS and VE-cadherin expression. Activation of the TLR4/MyD88/NF-κB and NLRP3 pathways may be responsible for PCSK9-induced endothelial dysfunction in sepsis. Vascular reactivity tests and survival studies showed that inhibition of PCSK9 could prevent the decrease in endothelium-dependent vasodilation function and improve the survival rates of septic mice. In summary, our results suggested that increased PCSK9 expression during sepsis activated the TLR4/MyD88/NF-κB and NLRP3 pathways to induce inflammation, which resulted in vascular endothelial dysfunction and decreased survival rates. Thus, inhibition of PCSK9 may be a potential clinical therapeutic target to improve vascular endothelial function in sepsis.
































 京公网安备 11010802027423号
京公网安备 11010802027423号