npj Computational Materials ( IF 9.4 ) Pub Date : 2022-08-02 , DOI: 10.1038/s41524-022-00846-z Shikha Saini , Joakim Halldin Stenlid , Frank Abild-Pedersen
|
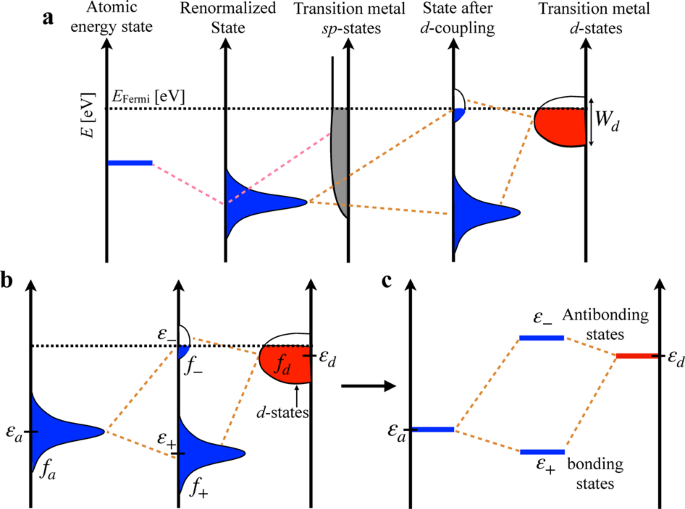
The chemisorption energy is an integral aspect of surface chemistry, central to numerous fields such as catalysis, corrosion, and nanotechnology. Electronic-structure-based methods such as the Newns-Anderson model are therefore of great importance in guiding the engineering of material surfaces with optimal properties. However, existing methods are inadequate for interpreting complex, multi-metallic systems. Herein, we introduce a physics-based chemisorption model for alloyed transition metal surfaces employing primarily metal d-band properties that accounts for perturbations in both the substrate and adsorbate electronic states upon interaction. Importantly, we show that adsorbate-induced changes in the adsorption site interact with its chemical environment leading to a second-order response in chemisorption energy with the d-filling of the neighboring atoms. We demonstrate the robustness of the model on a wide range of transition metal alloys with O, N, CH, and Li adsorbates yielding a mean absolute error of 0.13 eV versus density functional theory reference chemisorption energies.
中文翻译:
电子结构因素和吸附质效应在表面合金化学吸附中的重要性
化学吸附能是表面化学的一个组成部分,是催化、腐蚀和纳米技术等众多领域的核心。因此,基于电子结构的方法(例如 Newns-Anderson 模型)对于指导具有最佳性能的材料表面的工程设计非常重要。然而,现有的方法不足以解释复杂的多金属系统。在这里,我们介绍了一种基于物理的化学吸附模型,用于合金化过渡金属表面,主要采用金属d带特性,它解释了相互作用时基板和吸附电子态的扰动。重要的是,我们表明吸附质诱导的吸附位点变化与其化学环境相互作用,导致化学吸附能量的二级响应与相邻原子的d填充。我们证明了该模型在具有 O、N、CH 和 Li 吸附物的各种过渡金属合金上的稳健性,相对于密度泛函理论参考化学吸附能,产生 0.13 eV 的平均绝对误差。






























 京公网安备 11010802027423号
京公网安备 11010802027423号