Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2022-06-14 , DOI: 10.1016/j.bioorg.2022.105964 Zahra M Alamshany 1 , Nada Y Tashkandi 1 , Ismail M M Othman 2 , Manal M Anwar 3 , Eman S Nossier 4
|
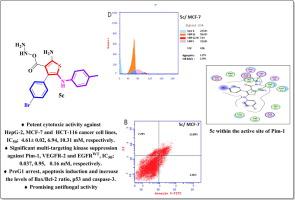
Multitargeting kinase inhibitors recently proved to be a profitable approach for conquering cancer proliferation. The current study represents the design and synthesis of new thiophene, thienopyridine, and thiazoline-based derivatives 4-14a,b. All the target compounds were examined in vitro against three cancer cell lines; the liver (HepG-2), breast (MCF-7), and colon (HCT-116) where the thiophene-based compounds 5a-c, demonstrated the most potent activity. Furthermore, the latter derivatives revealed a safety profile against WI-38 normal cell line of selectivity indices ranging from 4.43 to 17.44. In vitro enzyme assay of 5a-c revealed that the carbohydrazide analog 5c has the most promising multitargeting inhibiting activity against Pim-1, VEGFR-2, and EGFRWT enzymes of IC50 values; 0.037 ± 0.02, 0.95 ± 0.24, and 0.16 ± 0.05 µM, respectively. As it was the most potent analog, 5c was further subjected to cell cycle and apoptosis analysis. The results indicated that it induced preG1 arrest and an apoptotic effect in the early and late stages. Moreover, further apoptosis studies were carried out for 5c to evaluate its proapoptotic potential. Interestingly, 5c enhanced the levels of Bax/Bcl-2 ratio, p53, and active caspase 3 by 18, 6.4, and 24 folds, respectively compared to the untreated cells. The antimicrobial evaluation showed that only compounds 3 and 5a produced broad-spectrum potency, while 5b and 5c exhibited outstanding antifungal effects. Finally, a molecular docking study was carried out to discover the probable interactions of compound 5c with the active sites of Pim-1, VEGFR-2, and EGFRWT kinases.
中文翻译:
新型噻吩、噻吩并吡啶和噻唑啉衍生物:作为抗增殖剂和多靶点激酶抑制剂的设计、合成和生物学评价
多靶点激酶抑制剂最近被证明是征服癌症扩散的一种有利可图的方法。目前的研究代表了新型噻吩、噻吩并吡啶和噻唑啉基衍生物4-14a,b的设计和合成。所有目标化合物均在体外针对三种癌细胞系进行了检测;在肝脏 (HepG-2)、乳房 (MCF-7) 和结肠 (HCT-116) 中,噻吩基化合物5a-c表现出最有效的活性。此外,后一种衍生物揭示了针对 WI-38 正常细胞系的安全性,选择性指数范围为 4.43 至 17.44。5a-c的体外酶测定表明,碳酰肼类似物5c对 Pim-1、VEGFR-2 和 EGFR WT酶具有最有希望的多靶点抑制活性,IC 50值;分别为 0.037 ± 0.02、0.95 ± 0.24 和 0.16 ± 0.05 µM。由于它是最有效的类似物,因此对5c进行了进一步的细胞周期和细胞凋亡分析。结果表明它在早期和晚期诱导preG1停滞和凋亡作用。此外,对5c进行了进一步的凋亡研究以评估其促凋亡潜力。有趣的是,与未处理的细胞相比,5c将 Bax/Bcl-2 比率、p53 和活性 caspase 3 的水平分别提高了 18、6.4 和 24 倍。抗菌评估表明只有化合物35a和5a产生广谱效力,而5b和5c表现出显着的抗真菌作用。最后,进行分子对接研究以发现化合物5c与 Pim-1、VEGFR-2 和 EGFR WT激酶活性位点的可能相互作用。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号