当前位置:
X-MOL 学术
›
Nat. Commun.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
使用全基因组测序稳健而准确地估计重复基因的旁系同源特异性拷贝数
Nature Communications ( IF 14.7 ) Pub Date : 2022-06-09 , DOI: 10.1038/s41467-022-30930-3 Timofey Prodanov 1 , Vikas Bansal 2
Affiliation
|
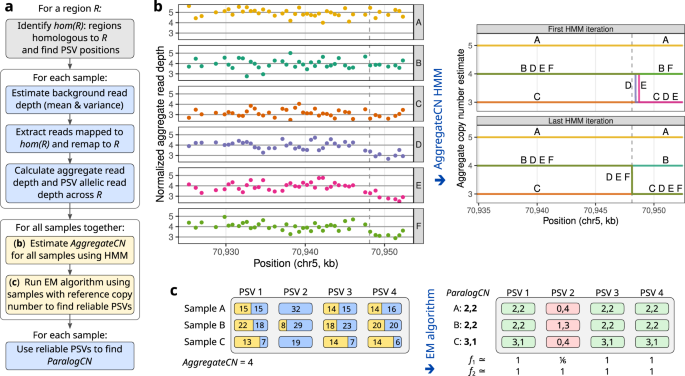
人类基因组包含数百个低拷贝重复序列 (LCR),由于广泛的拷贝数变异和读长映射的模糊性,使用短读长测序技术进行分析具有挑战性。超过 150 个与 LCR 重叠的重复基因的拷贝数和序列变异与单基因和复杂的人类疾病有关。我们描述了一种计算工具 Parascopy,用于使用全基因组测序 (WGS) 来估计重复基因的聚合和旁系同源特异性拷贝数。 Parascopy 是一种有效的方法,可以联合分析映射到不同重复副本的读取,而不需要全局重新对齐。它利用多个样本来减轻测序偏差并识别区分重复拷贝的可靠旁系同源序列变体 (PSV)。对来自不同人群的 2504 名个体的 WGS 数据分析表明,Parascopy 对测序偏差具有鲁棒性,与现有方法相比具有更高的准确性,并且能够优先考虑重复基因中的致病性拷贝数变化。
"点击查看英文标题和摘要"




















































 京公网安备 11010802027423号
京公网安备 11010802027423号