当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Kinetic Evidence of Most Abundant Surface Intermediates Variation over Ptn and Ptp: Few-Atom Pt Ensembles Enable Efficient Catalytic Cyclohexane Dehydrogenation for Hydrogen Production-II
ACS Catalysis ( IF 11.3 ) Pub Date : 2022-06-02 , DOI: 10.1021/acscatal.2c01420 Jinqiu Guo 1, 2 , Mi Peng 3 , Zhimin Jia 4, 5 , Chengyu Li 3 , Hongyang Liu 4, 5 , Hongbo Zhang 1, 2 , Ding Ma 3
ACS Catalysis ( IF 11.3 ) Pub Date : 2022-06-02 , DOI: 10.1021/acscatal.2c01420 Jinqiu Guo 1, 2 , Mi Peng 3 , Zhimin Jia 4, 5 , Chengyu Li 3 , Hongyang Liu 4, 5 , Hongbo Zhang 1, 2 , Ding Ma 3
Affiliation
|
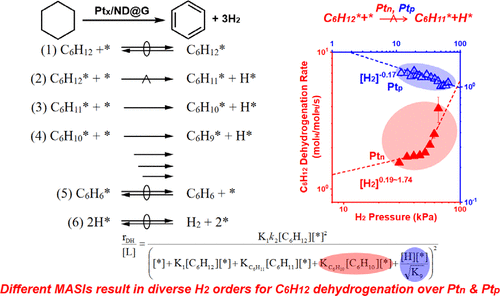
C–H bond activation is very important in upgrading of alkanes or aromatics into value-added products, such as alkenes or even oxygenates, etc., during natural gas utilization and hydrogen transportation via chemical strategies. Because of the low polarization ability and high bond energies of the C–H bonds within the hydrocarbons, the selective C–H bond activation suffers from low efficiency for a long time and requests systematic work on understanding the reaction mechanisms, in which the kinetic studies are highly desired and not well understood in a comprehensive manner. Herein, a universal kinetic model has been established to explain the entire reaction process of C–H bond activation for cyclohexane dehydrogenation (CDH) over various structures of Pt (cluster: Ptn and particle: Ptp) decorated nanodiamond@graphene (Ptx/ND@G) nanocomposite with specific emphasis on the elementary steps, the rate-determining step(s) (RDS), and the most abundant surface intermediates (MASIs). With the combination of kinetic and thermodynamic measurements, it was found that cyclohexane dehydrogenation shows different reaction mechanisms over Ptn catalysts, compared to Ptp catalysts. And C6H12 dehydrogenation rates showed nearly first-order (Ptn: rDH ∼ [C6H12]∼0.6; Ptp: rDH ∼ [C6H12]∼1.0) dependence on the C6H12, while the orders of H2 are obviously different over the two catalysts, in which the addition of H2 shows negligible effect (nearly zero-order) once a trace amount of H2 is introduced, and then obviously promotes (nearly second-order) dehydrogenation over Ptn (Ptn: rDH ∼ [H2]∼0–2), while the dehydrogenation rate over Ptp is prominently inhibited by adding H2 to the catalytic system under similar reaction conditions (Ptp: rDH ∼ [H2]∼ –1.0–0), thus indicating that Ptn and Ptp catalysts have diverse MASIs, which is further supported by the developed universal kinetic model of dehydrogenation. That perfectly explains the relationship between the dehydrogenation events and the coverage of surface hydrogen species. The predicted reaction process following a Langmuir–Hinshelwood model that matches the experimental configurations very well, suggesting that the first C–H bond rupture of C6H12 is probably the RDS for both Ptn and Ptp catalysts, while the MASIs varied from C6H10* to H* explained the diversities of the H2 dependencies between Ptn and Ptp catalysts systematically. This kinetic case study as well as the established universal model could be easily extended to some other systems related to C–H bond rupture and attracts the attention on exploring the correlations between nanostructure and the reaction performance.
中文翻译:

最丰富的表面中间体在 Ptn 和 Ptp 上变化的动力学证据:少原子 Pt 集合实现高效催化环己烷脱氢制氢-II
在天然气利用和通过化学策略运输氢气的过程中,C-H键活化对于将烷烃或芳烃升级为烯烃甚至含氧化合物等增值产品非常重要。由于烃类中 C-H 键的低极化能力和高键能,选择性 C-H 键活化长期以来效率低下,需要系统地研究反应机理,其中动力学研究非常需要并且不能以全面的方式很好地理解。在此,建立了一个通用动力学模型来解释环己烷脱氢(CDH)在各种 Pt 结构(簇:Pt n和颗粒:Pt p )上的 C-H 键活化的整个反应过程。) 修饰的纳米金刚石@石墨烯 (Pt x /ND@G) 纳米复合材料,特别强调基本步骤、速率决定步骤 (RDS) 和最丰富的表面中间体 (MASI)。通过结合动力学和热力学测量,发现与 Pt p催化剂相比,环己烷脱氢在 Pt n催化剂上表现出不同的反应机理。C 6 H 12脱氢率接近一级(Pt n : r DH ∼ [C 6 H 12 ] ∼0.6 ; Pt p : r DH ∼ [C 6 H12 ] ∼1.0 ) 对C 6 H 12的依赖性,而H 2的级数在两种催化剂上明显不同,其中H 2 的添加在微量H 2时表现出可忽略不计的效果(接近零级)被引入,然后明显促进 Pt n (Pt n : r DH ∼ [H 2 ] ∼0-2 ) 上的(接近二级)脱氢,而在 Pt p上的脱氢速率通过添加 H 2显着抑制类似反应条件下的催化体系(Pt p: r DH ∼ [H 2 ] ∼ –1.0-0 ),因此表明 Pt n和 Pt p催化剂具有不同的 MASI,这得到了开发的通用脱氢动力学模型的进一步支持。这完美地解释了脱氢事件与表面氢物种覆盖率之间的关系。根据 Langmuir-Hinshelwood 模型预测的反应过程与实验配置非常匹配,这表明 C 6 H 12的第一个 C-H 键断裂可能是 Pt n和 Pt p催化剂的 RDS,而 MASI 变化C 6 H 10* to H* 系统地解释了Pt n和Pt p催化剂之间H 2依赖性的多样性。该动力学案例研究以及所建立的通用模型可以很容易地扩展到与 C-H 键断裂相关的其他一些系统,并引起人们对探索纳米结构与反应性能之间相关性的关注。
更新日期:2022-06-02
中文翻译:
最丰富的表面中间体在 Ptn 和 Ptp 上变化的动力学证据:少原子 Pt 集合实现高效催化环己烷脱氢制氢-II
在天然气利用和通过化学策略运输氢气的过程中,C-H键活化对于将烷烃或芳烃升级为烯烃甚至含氧化合物等增值产品非常重要。由于烃类中 C-H 键的低极化能力和高键能,选择性 C-H 键活化长期以来效率低下,需要系统地研究反应机理,其中动力学研究非常需要并且不能以全面的方式很好地理解。在此,建立了一个通用动力学模型来解释环己烷脱氢(CDH)在各种 Pt 结构(簇:Pt n和颗粒:Pt p )上的 C-H 键活化的整个反应过程。) 修饰的纳米金刚石@石墨烯 (Pt x /ND@G) 纳米复合材料,特别强调基本步骤、速率决定步骤 (RDS) 和最丰富的表面中间体 (MASI)。通过结合动力学和热力学测量,发现与 Pt p催化剂相比,环己烷脱氢在 Pt n催化剂上表现出不同的反应机理。C 6 H 12脱氢率接近一级(Pt n : r DH ∼ [C 6 H 12 ] ∼0.6 ; Pt p : r DH ∼ [C 6 H12 ] ∼1.0 ) 对C 6 H 12的依赖性,而H 2的级数在两种催化剂上明显不同,其中H 2 的添加在微量H 2时表现出可忽略不计的效果(接近零级)被引入,然后明显促进 Pt n (Pt n : r DH ∼ [H 2 ] ∼0-2 ) 上的(接近二级)脱氢,而在 Pt p上的脱氢速率通过添加 H 2显着抑制类似反应条件下的催化体系(Pt p: r DH ∼ [H 2 ] ∼ –1.0-0 ),因此表明 Pt n和 Pt p催化剂具有不同的 MASI,这得到了开发的通用脱氢动力学模型的进一步支持。这完美地解释了脱氢事件与表面氢物种覆盖率之间的关系。根据 Langmuir-Hinshelwood 模型预测的反应过程与实验配置非常匹配,这表明 C 6 H 12的第一个 C-H 键断裂可能是 Pt n和 Pt p催化剂的 RDS,而 MASI 变化C 6 H 10* to H* 系统地解释了Pt n和Pt p催化剂之间H 2依赖性的多样性。该动力学案例研究以及所建立的通用模型可以很容易地扩展到与 C-H 键断裂相关的其他一些系统,并引起人们对探索纳米结构与反应性能之间相关性的关注。





























 京公网安备 11010802027423号
京公网安备 11010802027423号