当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adsorption properties of pyramidal superatomic molecules based on the structural framework of the Au20 cluster
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-05-02 , DOI: 10.1039/d2cp01552h
Qiman Liu 1, 2 , Manli Zhang 1 , Dawen Zhang 1 , Yunhu Hu 1 , Qiyong Zhu 1 , Longjiu Cheng 2
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-05-02 , DOI: 10.1039/d2cp01552h
Qiman Liu 1, 2 , Manli Zhang 1 , Dawen Zhang 1 , Yunhu Hu 1 , Qiyong Zhu 1 , Longjiu Cheng 2
Affiliation
|
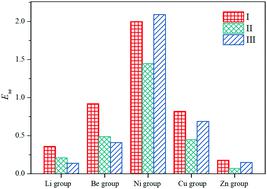
The pyramidal Au20 cluster is a highly inert and stable superatomic molecule, but it is not suitable as a potential catalyst for covalent bond activations, e.g., CO oxidation reaction. Herein, the adsorption and electronic properties of CO molecules on various pyramidal clusters based on the structural framework of Au20 are investigated using density functional theory. According to the SVB model, we constructed isoelectronic superatomic molecules with different pyramid configurations by replacing the vertex atoms of the Au20 using metal M atoms (M = Li, Be, Ni, Cu, and Zn group atoms). After the CO molecules are adsorbed on the vertex atoms of these metal clusters, we analyzed the CO adsorption energies, C–O bond stretching frequencies, and electronic properties of the adsorption structures. It was found that the adsorption of CO molecules results in minimal changes in the parent geometries of the pyramidal clusters, and most adsorption structures are consistent with the geometry of CO adsorption at the vertex site of the Au20 cluster. There are significant red shifts when CO molecules are adsorbed on the Ni/Pd/Pt atoms of the clusters, and their CO adsorption energies were also greater. The molecular orbitals and density of states reveal that there are overlaps between the frontier orbitals of the clusters and CO, and the electronic structure of NiAu19− is not sensitive to CO. The ETS-NOCV analysis shows that the increase in the density of the bonding area caused by the orbital interactions between the fragments is higher than the decrease in the density of the bonding area caused by Pauli repulsion, presenting that the direction of charge flow in the deformation density is from CO → clusters. From energy decomposition analysis (EDA) and NPA charge, we find a predominant covalent nature of the contributions in CO⋯M interactions (σ-donation). Our study indicates that the SVB model provides a new direction to expand the superatomic catalysts from the superatom clusters, which also provides inference for the extension of the single atom catalysis.
中文翻译:

基于Au20簇结构框架的锥体超原子分子吸附特性
锥体Au 20簇是一种高度惰性和稳定的超原子分子,但它不适合作为共价键活化的潜在催化剂,例如CO 氧化反应。在此,基于Au 20的结构框架,利用密度泛函理论研究了CO分子在各种锥体簇上的吸附和电子性质。根据SVB模型,我们通过替换Au 20的顶点原子构建了具有不同金字塔构型的等电子超原子分子。使用金属 M 原子(M = Li、Be、Ni、Cu 和 Zn 基团原子)。在 CO 分子吸附在这些金属簇的顶点原子上后,我们分析了 CO 吸附能、C-O 键拉伸频率和吸附结构的电子特性。发现CO分子的吸附导致锥体簇的母体几何形状发生微小变化,并且大多数吸附结构与Au 20顶点位置的CO吸附几何形状一致簇。当CO分子吸附在团簇的Ni/Pd/Pt原子上时存在明显的红移,并且它们的CO吸附能也更大。分子轨道和状态密度表明,团簇的前沿轨道和 CO 之间存在重叠,以及 NiAu 19的电子结构-对CO不敏感。ETS-NOCV分析表明,碎片间轨道相互作用引起的键合区密度增加大于泡利斥力引起的键合区密度降低,表明变形密度中的电荷流方向来自CO→簇。从能量分解分析 (EDA) 和 NPA 电荷中,我们发现在 CO⋯M 相互作用(σ-捐赠)中贡献的主要共价性质。我们的研究表明,SVB模型为从超原子团簇中扩展超原子催化剂提供了新的方向,也为单原子催化的扩展提供了推论。
更新日期:2022-05-02
中文翻译:
基于Au20簇结构框架的锥体超原子分子吸附特性
锥体Au 20簇是一种高度惰性和稳定的超原子分子,但它不适合作为共价键活化的潜在催化剂,例如CO 氧化反应。在此,基于Au 20的结构框架,利用密度泛函理论研究了CO分子在各种锥体簇上的吸附和电子性质。根据SVB模型,我们通过替换Au 20的顶点原子构建了具有不同金字塔构型的等电子超原子分子。使用金属 M 原子(M = Li、Be、Ni、Cu 和 Zn 基团原子)。在 CO 分子吸附在这些金属簇的顶点原子上后,我们分析了 CO 吸附能、C-O 键拉伸频率和吸附结构的电子特性。发现CO分子的吸附导致锥体簇的母体几何形状发生微小变化,并且大多数吸附结构与Au 20顶点位置的CO吸附几何形状一致簇。当CO分子吸附在团簇的Ni/Pd/Pt原子上时存在明显的红移,并且它们的CO吸附能也更大。分子轨道和状态密度表明,团簇的前沿轨道和 CO 之间存在重叠,以及 NiAu 19的电子结构-对CO不敏感。ETS-NOCV分析表明,碎片间轨道相互作用引起的键合区密度增加大于泡利斥力引起的键合区密度降低,表明变形密度中的电荷流方向来自CO→簇。从能量分解分析 (EDA) 和 NPA 电荷中,我们发现在 CO⋯M 相互作用(σ-捐赠)中贡献的主要共价性质。我们的研究表明,SVB模型为从超原子团簇中扩展超原子催化剂提供了新的方向,也为单原子催化的扩展提供了推论。
































 京公网安备 11010802027423号
京公网安备 11010802027423号