当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
用线性响应驱动的分子动力学计算蛋白质配体结合的构象转变途径
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-04-28 , DOI: 10.1021/acs.jctc.1c01243 Rajat Punia 1 , Gaurav Goel 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-04-28 , DOI: 10.1021/acs.jctc.1c01243 Rajat Punia 1 , Gaurav Goel 1
Affiliation
|
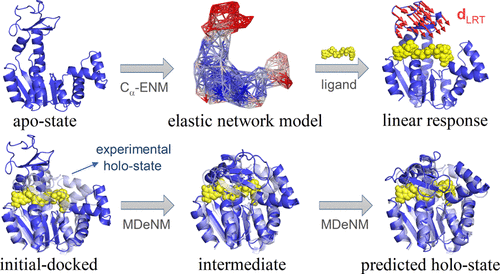
虽然对于将蛋白质结构与其生物学功能联系起来极为重要,但由于中间体的瞬态性、大而崎岖的构象空间以及蛋白质动力学和配体-蛋白质之间的耦合,很难确定配体结合后的蛋白质构象转变途径互动。依赖于绑定(全息)状态结构的先验知识的现有方法是限制性的。第二个关注点与获得的中间体与 apo → holo 过渡途径上的亚稳态的对应关系有关。这里,我们将蛋白质 apo 结构和配体结合位点作为唯一输入,并将蛋白质哈密顿量的弹性网络模型 (ENM) 表示与蛋白质 - 配体相互作用的线性响应理论 (LRT) 相结合,以确定一组慢速正常模式与蛋白质构象变化方向高度重叠的蛋白质振动。沿所选方向的结构位移是使用激发的正常模式分子动力学 (MDeNM) 模拟而不是直接使用 LRT 进行的。在此,基于其与蛋白质动力学和配体-蛋白质相互作用的耦合,MDeNM 激发速度被即时优化。因此,针对四种蛋白质-配体系统的晶体学和模拟数据验证了一组确定的结构,即,d-吡喃核糖、DNA β-葡萄糖基转移酶-尿苷-5'-二磷酸和 G-蛋白 α 亚基-鸟苷-5'-三磷酸,它们在蛋白质构象异质性、配体结合机制等方面存在重要差异。配体结合后蛋白质构象变化的诱导拟合或构象选择、程度和非线性,以及变构效应的存在。获得的一组中间体用作路径元动力学模拟的输入,以获得 apo → 全息跃迁的自由能分布。

"点击查看英文标题和摘要"
更新日期:2022-04-28
"点击查看英文标题和摘要"





























 京公网安备 11010802027423号
京公网安备 11010802027423号