Journal of Superconductivity and Novel Magnetism ( IF 1.6 ) Pub Date : 2022-04-08 , DOI: 10.1007/s10948-022-06239-z
A. Harbi 1 , S. Benmokhtar 1 , M. Moutaabbid 1 , A. Azouaoui 2
|
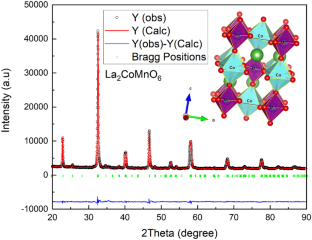
La2CoMnO6 double perovskite was synthesized by the solid-state reaction method, and its crystal structure was investigated using X-ray diffraction and Raman spectroscopy. The crystal structure of the study compound has been verified to possess monoclinic crystal symmetry, space group P21/n with the lattice parameters a = 5.53 Å, b = 5.48 Å, c = 7.77 Å, and β = 89.9°. The Raman spectrum shows two broad peaks, the high-frequency peak near 668 cm−1 corresponding to the Ag stretching mode, whereas the low-frequency peak near 522 cm−1 corresponding to the Bg anti-stretching and bending modes. The electronic and magnetic properties of pure and defective perovskite La2CoMnO6 have been carried out using the density functional theory (DFT). The result shows a ferromagnetic insulator character for pure perovskite; the ferromagnetic state is explained by super-exchange interaction between empty and half-filled eg orbitals of Mn4+( \({t}_{2g}^{3}\) \({e}_{g}^{0}\)) and Co2+ (\({t}_{2g}^{5}{e}_{g}^{2}\)) via oxygen O2−. The valence band maximum (VBM) begins primarily from the p-states of oxygen atom and d-states of transition metal (Mn and Co), while the conduction band minimum (CBM) comprises both d-states of the M-site transition metal and f-state of lanthanum. The creation of oxygen defect leads to the lattice expansion due to a Coulomb repulsion between the Co atom and Mn atom as oxygen is no longer their nearest neighbor (NN). The oxygen vacancy also reveals a half metallicity character with a bandgap in the spin-up channel and a continuous band at the Fermi level in the spin-down channel. The calculated Curie temperature TC decreased (pure Tc = 422 K and defective Tc = 378 K) as decreasing the bond angle Mn-\(\widehat{O}\) 3-Co (pure Mn-\(\widehat{O}\) 3-Co = 150.13° and defective Mn-\(\widehat{O}\) 3-Co = 146.026°).
中文翻译:
氧空位对 La2CoMnO6 的结构、磁性和电子性质的作用的合成、表征和 DFT 研究
采用固相反应法合成了La 2 CoMnO 6双钙钛矿,并利用X射线衍射和拉曼光谱对其晶体结构进行了研究。研究化合物的晶体结构经证实具有单斜晶体对称性,空间群 P2 1 /n,晶格参数a = 5.53 Å,b = 5.48 Å,c = 7.77 Å,β = 89.9°。拉曼光谱显示两个宽峰,668 cm -1附近的高频峰对应于A g拉伸模式,而522 cm -1附近的低频峰对应于B g抗拉伸和弯曲模式。使用密度泛函理论 (DFT)对纯和有缺陷的钙钛矿 La 2 CoMnO 6的电子和磁性特性进行了研究。结果显示了纯钙钛矿的铁磁绝缘体特性;铁磁状态可以通过Mn 4+ ( \({t}_{2g}^{3}\) \ ({e}_{g}^{ 0}\) ) 和 Co 2+ ( \({t}_{2g}^{5}{e}_{g}^{2}\) ) 通过氧气 O 2− . 价带最大值 (VBM) 主要从氧原子的 p 态和过渡金属 (Mn 和 Co) 的 d 态开始,而导带最小值 (CBM) 包括 M 位过渡金属的两个 d 态和镧的f态。由于 Co 原子和 Mn 原子之间的库仑排斥,氧缺陷的产生导致晶格膨胀,因为氧不再是它们的最近邻 (NN)。氧空位还显示出半金属丰度特征,在自旋通道中具有带隙,在自旋通道中的费米能级处具有连续带。 随着键角 Mn- 的减小,计算出的居里温度T C降低(纯T c = 422 K 和缺陷T c = 378 K)\(\widehat{O}\) 3 -Co (纯 Mn- \(\widehat{O}\) 3 -Co = 150.13° 和有缺陷的 Mn- \(\widehat{O}\) 3 -Co = 146.026° )。
































 京公网安备 11010802027423号
京公网安备 11010802027423号