当前位置:
X-MOL 学术
›
Mol. Pharmaceutics
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Drug Conjugation via Maleimide–Thiol Chemistry Does Not Affect Targeting Properties of Cysteine-Containing Anti-FGFR1 Peptibodies
Molecular Pharmaceutics ( IF 4.5 ) Pub Date : 2022-04-07 , DOI: 10.1021/acs.molpharmaceut.1c00946 Karolina Jendryczko 1 , Jakub Rzeszotko 1 , Mateusz Adam Krzyscik 1 , Anna Kocyła 2 , Jakub Szymczyk 1 , Jacek Otlewski 1 , Anna Szlachcic 1
Molecular Pharmaceutics ( IF 4.5 ) Pub Date : 2022-04-07 , DOI: 10.1021/acs.molpharmaceut.1c00946 Karolina Jendryczko 1 , Jakub Rzeszotko 1 , Mateusz Adam Krzyscik 1 , Anna Kocyła 2 , Jakub Szymczyk 1 , Jacek Otlewski 1 , Anna Szlachcic 1
Affiliation
|
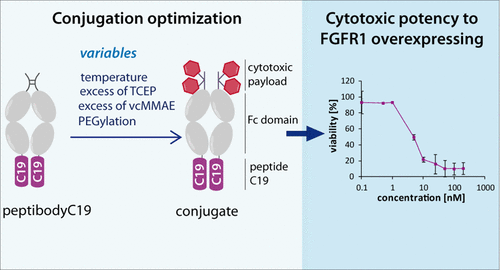
With a wide range of available cytotoxic therapeutics, the main focus of current cancer research is to deliver them specifically to the cancer cells, minimizing toxicity against healthy tissues. Targeted therapy utilizes different carriers for cytotoxic drugs, combining a targeting molecule, typically an antibody, and a highly toxic payload. For the effective delivery of such cytotoxic conjugates, a molecular target on the cancer cell is required. Various proteins are exclusively or abundantly expressed in cancer cells, making them a possible target for drug carriers. Fibroblast growth factor receptor 1 (FGFR1) overexpression has been reported in different types of cancer, but no FGFR1-targeting cytotoxic conjugate has been approved for therapy so far. In this study, the FGFR1-targeting peptide previously described in the literature was reformatted into a peptibody–peptide fusion with the fragment crystallizable (Fc) domain of IgG1. PeptibodyC19 can be effectively internalized into FGFR1-overexpressing cells and does not induce cells’ proliferation. The main challenge for its use as a cytotoxic conjugate is a cysteine residue located within the targeting peptide. A standard drug-conjugation strategy based on the maleimide–thiol reaction involves modification of cysteines within the Fc domain hinge region. Applied here, however, may easily result in the modification of the targeting peptide with the drug, limiting its affinity to the target and therefore the potential for specific drug delivery. To investigate if this is the case, we have performed conjugation reactions with different auristatin derivatives (PEGylated and unmodified) under various conditions. By controlling the reduction conditions and the type of cytotoxic payload, different numbers of cysteines were substituted, allowing us to avoid conjugating the drug to the targeting peptide, which could affect its binding to FGFR1. The optimized protocol with PEGylated auristatin yielded doubly substituted peptibodyC19, showing specific cytotoxicity toward the FGFR1-expressing lung cancer cells, with no effect on cells with low FGFR1 levels. Indeed, additional cysteine poses a risk of unwanted modification, but changes in the type of cytotoxic payload and reaction conditions allow the use of standard thiol–maleimide-based conjugation to achieve standard Fc hinge region cysteine modification, analogously to antibody–drug conjugates.
中文翻译:

通过马来酰亚胺-硫醇化学进行的药物缀合不会影响含半胱氨酸的抗 FGFR1 肽体的靶向特性
凭借多种可用的细胞毒性疗法,当前癌症研究的主要焦点是将它们特异性地递送至癌细胞,从而最大限度地减少对健康组织的毒性。靶向治疗利用不同的细胞毒性药物载体,结合靶向分子(通常是抗体)和高毒性有效负载。为了有效递送此类细胞毒性缀合物,需要癌细胞上的分子靶点。各种蛋白质在癌细胞中专门或大量表达,使它们成为药物载体的可能靶点。据报道,成纤维细胞生长因子受体 1 (FGFR1) 在不同类型的癌症中过度表达,但迄今为止尚未批准用于治疗的 FGFR1 靶向细胞毒性偶联物。在这项研究中,之前文献中描述的 FGFR1 靶向肽被重新格式化为具有 IgG1 片段可结晶 (Fc) 结构域的肽体-肽融合物。 PeptibodyC19可以有效地内化到FGFR1过表达的细胞中,并且不会诱导细胞增殖。其用作细胞毒性缀合物的主要挑战是位于靶向肽内的半胱氨酸残基。基于马来酰亚胺-硫醇反应的标准药物缀合策略涉及 Fc 结构域铰链区内半胱氨酸的修饰。然而,此处应用可能很容易导致靶向肽被药物修饰,限制其与靶点的亲和力,从而限制特异性药物递送的潜力。为了研究情况是否如此,我们在各种条件下与不同的阿里他汀衍生物(聚乙二醇化和未修饰的)进行了缀合反应。 通过控制还原条件和细胞毒性有效负载的类型,取代了不同数量的半胱氨酸,使我们能够避免药物与靶向肽缀合,这可能会影响其与 FGFR1 的结合。使用聚乙二醇化 auristatin 的优化方案产生了双取代肽体 C19,对表达 FGFR1 的肺癌细胞表现出特异性细胞毒性,但对 FGFR1 水平低的细胞没有影响。事实上,额外的半胱氨酸会带来不必要的修饰的风险,但细胞毒性有效负载类型和反应条件的变化允许使用标准的硫醇-马来酰亚胺缀合来实现标准的 Fc 铰链区半胱氨酸修饰,类似于抗体-药物缀合物。
更新日期:2022-04-07
中文翻译:
通过马来酰亚胺-硫醇化学进行的药物缀合不会影响含半胱氨酸的抗 FGFR1 肽体的靶向特性
凭借多种可用的细胞毒性疗法,当前癌症研究的主要焦点是将它们特异性地递送至癌细胞,从而最大限度地减少对健康组织的毒性。靶向治疗利用不同的细胞毒性药物载体,结合靶向分子(通常是抗体)和高毒性有效负载。为了有效递送此类细胞毒性缀合物,需要癌细胞上的分子靶点。各种蛋白质在癌细胞中专门或大量表达,使它们成为药物载体的可能靶点。据报道,成纤维细胞生长因子受体 1 (FGFR1) 在不同类型的癌症中过度表达,但迄今为止尚未批准用于治疗的 FGFR1 靶向细胞毒性偶联物。在这项研究中,之前文献中描述的 FGFR1 靶向肽被重新格式化为具有 IgG1 片段可结晶 (Fc) 结构域的肽体-肽融合物。 PeptibodyC19可以有效地内化到FGFR1过表达的细胞中,并且不会诱导细胞增殖。其用作细胞毒性缀合物的主要挑战是位于靶向肽内的半胱氨酸残基。基于马来酰亚胺-硫醇反应的标准药物缀合策略涉及 Fc 结构域铰链区内半胱氨酸的修饰。然而,此处应用可能很容易导致靶向肽被药物修饰,限制其与靶点的亲和力,从而限制特异性药物递送的潜力。为了研究情况是否如此,我们在各种条件下与不同的阿里他汀衍生物(聚乙二醇化和未修饰的)进行了缀合反应。 通过控制还原条件和细胞毒性有效负载的类型,取代了不同数量的半胱氨酸,使我们能够避免药物与靶向肽缀合,这可能会影响其与 FGFR1 的结合。使用聚乙二醇化 auristatin 的优化方案产生了双取代肽体 C19,对表达 FGFR1 的肺癌细胞表现出特异性细胞毒性,但对 FGFR1 水平低的细胞没有影响。事实上,额外的半胱氨酸会带来不必要的修饰的风险,但细胞毒性有效负载类型和反应条件的变化允许使用标准的硫醇-马来酰亚胺缀合来实现标准的 Fc 铰链区半胱氨酸修饰,类似于抗体-药物缀合物。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号