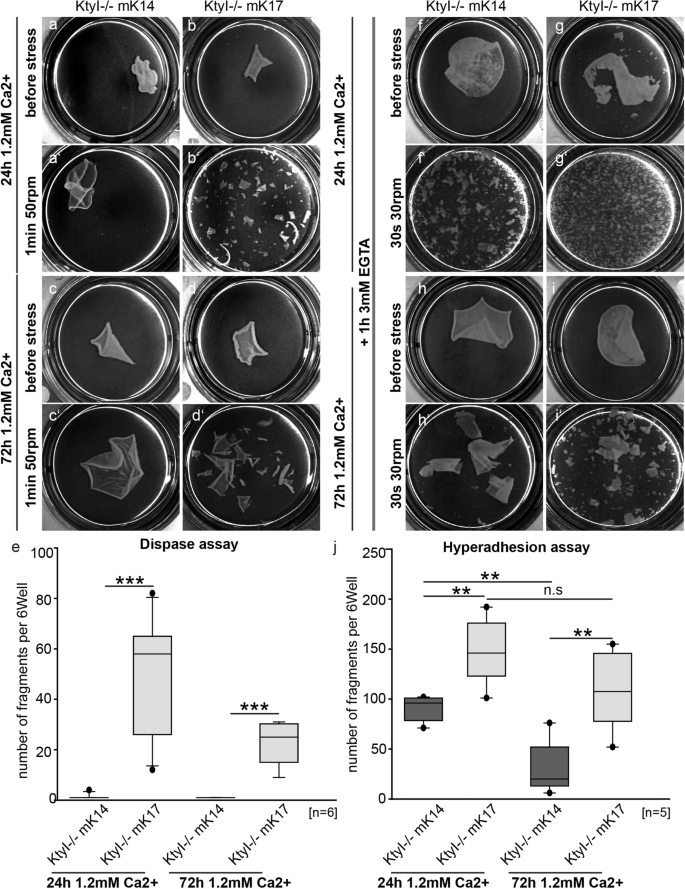
桥粒是细胞间连接,它通过将中间丝状细胞骨架束缚到质膜来介导暴露于机械应变的组织中的凝聚力和通讯。虽然成熟桥粒的特征在于过度粘附、Ca 2+非依赖性状态,但它们在伤口愈合、发病机制和组织再生过程中会暂时失去这种状态。控制过度粘附状态的机制仍未完全了解。在这里,我们表明在 Ca 2+- 诱导的角质形成细胞分化,角蛋白 17 (K17) 的表达阻止了稳定和过度粘附桥粒的形成,伴随着桥粒蛋白 (DP)、plakophilin-1 (PKP1)、desmoglein-1 (Dsg1) 和 -3 (Dsg3) 的显着减少) 在细胞间细胞边界。原子力显微镜显示,与表达 K14 的细胞相比,K17 中增加的桥粒芯核蛋白 3 分子的结合强度和桥粒芯核蛋白 3 寡聚体的量(已知的过度粘附的标志)都减少了。重要的是,Dsg3 或 DPII 的过表达增强了它们在细胞间细胞边界的定位并增加了 Dsg3 寡聚体的形成,尽管存在 K17,但仍会产生稳定的超粘附桥粒。值得注意的是,PKP1 在这些桥粒中富集。定量图像分析显示,DPII 过表达通过增加富含桥粒芯蛋白 3 的桥粒附近的 K5/K17 阳性角蛋白丝的丰度而导致桥粒过度粘附。因此,我们的数据表明,过度粘附可能是由于角蛋白同种型 K5/K17 募集到桥粒或由于 DP 和 Dsg3 的表达增强而与角蛋白组成无关。在没有角蛋白的情况下不能形成过度粘附桥粒的概念强调了角蛋白的重要作用,并在多个层面上提出了双向控制机制。我们的数据表明,过度粘附可能是由于将角蛋白同种型 K5/K17 募集到桥粒或由于 DP 和 Dsg3 的表达增强而与角蛋白组成无关。在没有角蛋白的情况下不能形成过度粘附桥粒的概念强调了角蛋白的重要作用,并在多个层面上提出了双向控制机制。我们的数据表明,过度粘附可能是由于将角蛋白同种型 K5/K17 募集到桥粒或由于 DP 和 Dsg3 的表达增强而与角蛋白组成无关。在没有角蛋白的情况下不能形成过度粘附桥粒的概念强调了角蛋白的重要作用,并在多个层面上提出了双向控制机制。
 "点击查看英文标题和摘要"
"点击查看英文标题和摘要"
Bidirectional regulation of desmosome hyperadhesion by keratin isotypes and desmosomal components
Desmosomes are intercellular junctions which mediate cohesion and communication in tissues exposed to mechanical strain by tethering the intermediate filament cytoskeleton to the plasma membrane. While mature desmosomes are characterized by a hyperadhesive, Ca2+-independent state, they transiently loose this state during wound healing, pathogenesis and tissue regeneration. The mechanisms controlling the hyperadhesive state remain incompletely understood. Here, we show that upon Ca2+-induced keratinocyte differentiation, expression of keratin 17 (K17) prevents the formation of stable and hyperadhesive desmosomes, accompanied by a significant reduction of desmoplakin (DP), plakophilin-1 (PKP1), desmoglein-1 (Dsg1) and -3 (Dsg3) at intercellular cell borders. Atomic force microscopy revealed that both increased binding strength of desmoglein-3 molecules and amount of desmoglein-3 oligomers, known hallmarks of hyperadhesion, were reduced in K17- compared to K14-expressing cells. Importantly, overexpression of Dsg3 or DPII enhanced their localization at intercellular cell borders and increased the formation of Dsg3 oligomers, resulting in stable, hyperadhesive desmosomes despite the presence of K17. Notably, PKP1 was enriched in these desmosomes. Quantitative image analysis revealed that DPII overexpression contributed to desmosome hyperadhesion by increasing the abundance of K5/K17-positive keratin filaments in the proximity of desmosomes enriched in desmoglein-3. Thus, our data show that hyperadhesion can result from recruitment of keratin isotypes K5/K17 to desmosomes or from enhanced expression of DP and Dsg3 irrespective of keratin composition. The notion that hyperadhesive desmosomes failed to form in the absence of keratins underscores the essential role of keratins and suggest bidirectional control mechanisms at several levels.






























 京公网安备 11010802027423号
京公网安备 11010802027423号