当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
GLOW:集成高斯加速分子动力学和深度学习的自由能分析工作流程
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-02-24 , DOI: 10.1021/acs.jctc.1c01055 Hung N Do 1 , Jinan Wang 1 , Apurba Bhattarai 1 , Yinglong Miao 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-02-24 , DOI: 10.1021/acs.jctc.1c01055 Hung N Do 1 , Jinan Wang 1 , Apurba Bhattarai 1 , Yinglong Miao 1
Affiliation
|
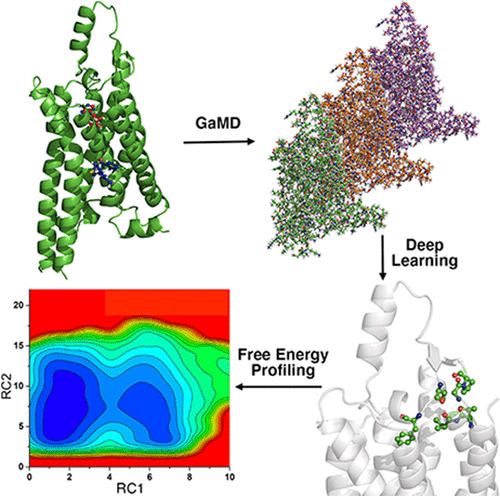
我们引入了高斯加速分子动力学 (GaMD)、深度学习 (DL) 和自由能分析工作流程 (GLOW) 来预测分子决定因素并绘制生物分子的自由能图谱。全原子 GaMD 增强采样模拟首先对感兴趣的生物分子进行。然后根据 GaMD 模拟帧计算结构接触图,并将其转换为图像,以使用卷积神经网络构建 DL 模型。重要的结构接触是根据结构接触梯度的注意力图的 DL 模型进一步确定的,这使我们能够识别系统反应坐标。最后,通过 GaMD 模拟的能量重新加权,为选定的反应坐标计算自由能分布。1受体 (A 1 AR) 作为模型系统。GLOW 的发现与之前对 A 1 AR的实验和计算研究高度一致,同时还提供了对受体功能的进一步机制见解。总之,GLOW 提供了一种系统的方法来绘制生物分子的自由能景观。GLOW 工作流程及其用户手册可在 http://miaolab.org/GLOW 下载。

"点击查看英文标题和摘要"
更新日期:2022-02-24
"点击查看英文标题和摘要"




















































 京公网安备 11010802027423号
京公网安备 11010802027423号