当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
GLOW: A Workflow Integrating Gaussian-Accelerated Molecular Dynamics and Deep Learning for Free Energy Profiling
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-02-24 , DOI: 10.1021/acs.jctc.1c01055
Hung N Do 1 , Jinan Wang 1 , Apurba Bhattarai 1 , Yinglong Miao 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2022-02-24 , DOI: 10.1021/acs.jctc.1c01055
Hung N Do 1 , Jinan Wang 1 , Apurba Bhattarai 1 , Yinglong Miao 1
Affiliation
|
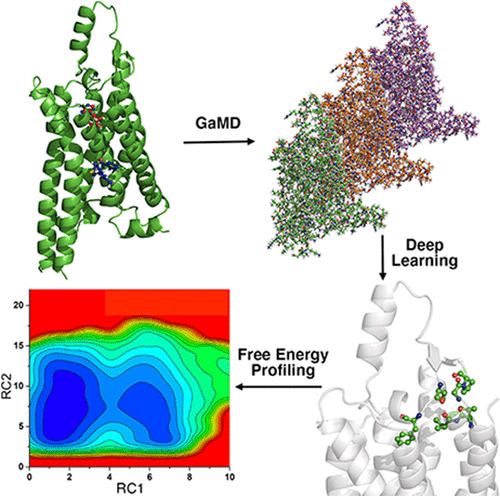
We introduce a Gaussian-accelerated molecular dynamics (GaMD), deep learning (DL), and free energy profiling workflow (GLOW) to predict molecular determinants and map free energy landscapes of biomolecules. All-atom GaMD-enhanced sampling simulations are first performed on biomolecules of interest. Structural contact maps are then calculated from GaMD simulation frames and transformed into images for building DL models using a convolutional neural network. Important structural contacts are further determined from DL models of attention maps of the structural contact gradients, which allow us to identify the system reaction coordinates. Finally, free energy profiles are calculated for the selected reaction coordinates through energetic reweighting of the GaMD simulations. We have also successfully demonstrated GLOW for the characterization of activation and allosteric modulation of a G protein-coupled receptor, using the adenosine A1 receptor (A1AR) as a model system. GLOW findings are highly consistent with previous experimental and computational studies of the A1AR, while also providing further mechanistic insights into the receptor function. In summary, GLOW provides a systematic approach to mapping free energy landscapes of biomolecules. The GLOW workflow and its user manual can be downloaded at http://miaolab.org/GLOW.
中文翻译:

GLOW:集成高斯加速分子动力学和深度学习的自由能分析工作流程
我们引入了高斯加速分子动力学 (GaMD)、深度学习 (DL) 和自由能分析工作流程 (GLOW) 来预测分子决定因素并绘制生物分子的自由能图谱。全原子 GaMD 增强采样模拟首先对感兴趣的生物分子进行。然后根据 GaMD 模拟帧计算结构接触图,并将其转换为图像,以使用卷积神经网络构建 DL 模型。重要的结构接触是根据结构接触梯度的注意力图的 DL 模型进一步确定的,这使我们能够识别系统反应坐标。最后,通过 GaMD 模拟的能量重新加权,为选定的反应坐标计算自由能分布。1受体 (A 1 AR) 作为模型系统。GLOW 的发现与之前对 A 1 AR的实验和计算研究高度一致,同时还提供了对受体功能的进一步机制见解。总之,GLOW 提供了一种系统的方法来绘制生物分子的自由能景观。GLOW 工作流程及其用户手册可在 http://miaolab.org/GLOW 下载。
更新日期:2022-02-24
中文翻译:
GLOW:集成高斯加速分子动力学和深度学习的自由能分析工作流程
我们引入了高斯加速分子动力学 (GaMD)、深度学习 (DL) 和自由能分析工作流程 (GLOW) 来预测分子决定因素并绘制生物分子的自由能图谱。全原子 GaMD 增强采样模拟首先对感兴趣的生物分子进行。然后根据 GaMD 模拟帧计算结构接触图,并将其转换为图像,以使用卷积神经网络构建 DL 模型。重要的结构接触是根据结构接触梯度的注意力图的 DL 模型进一步确定的,这使我们能够识别系统反应坐标。最后,通过 GaMD 模拟的能量重新加权,为选定的反应坐标计算自由能分布。1受体 (A 1 AR) 作为模型系统。GLOW 的发现与之前对 A 1 AR的实验和计算研究高度一致,同时还提供了对受体功能的进一步机制见解。总之,GLOW 提供了一种系统的方法来绘制生物分子的自由能景观。GLOW 工作流程及其用户手册可在 http://miaolab.org/GLOW 下载。
































 京公网安备 11010802027423号
京公网安备 11010802027423号