Chemical Engineering Journal ( IF 13.3 ) Pub Date : 2022-02-23 , DOI: 10.1016/j.cej.2022.135428 Jiahui Hu 1, 2 , Yin Li 1, 2 , Yubin Zou 1, 3 , Lin Lin 1, 2 , Bing Li 1, 2 , Xiao-yan Li 2, 3, 4
|
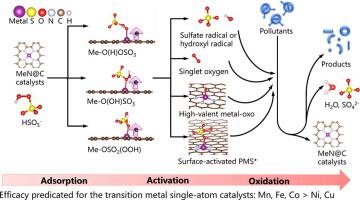
Single-atom catalysts perform excellently in peroxymonosulfate (PMS)-based advanced oxidation processes (AOPs), for which the generation of reactive oxygen species (ROS) is essential to the degradation of emerging organic pollutants in water. However, the detailed PMS activation mechanisms remain elusive. Density functional theory (DFT) calculation as a powerful approach can overcome the limitations of the experimental studies, providing a molecular-level perspective of catalytic process. This study conducted DFT calculations to clarify the electronic structures and PMS adsorption and activation mechanisms of a series of transition metal single-atom catalysts. According to the DFT study, significant electronic interaction and negative formation energy make nitrogen-doped carbon (N@C) supports suitable for stabilizing metal atoms (Me) to form MeN@C catalysts. As the active site, single metal atom adsorbs the oxygen atoms of PMS by electrostatic and magnetic interactions, and transfer electrons from MeN@C to activate PMS. Different adsorption configurations and the subsequent PMS activation lead to the generation of various ROS, including the SO4•- radical, •OH radical, singlet oxygen (1O2), high-valent metal-oxo species, and surface-activated PMS*. Electron transfer mediated by surface-activated PMS* may dominate in all MeN@C/PMS systems. The generation of free radicals can be difficult for some systems. High-valent metal-oxo species are readily formed by FeN@C/PMS and MnN@C/PMS, whereas 1O2 tends to be produced by CoN@C/PMS, NiN@C/PMS, and CuN@C/PMS. The findings will provide a theoretical basis for the design and synthesis of effective MeN@C catalysts for the AOPs to remove emerging organic pollutants from water and wastewater.
中文翻译:
嵌入 N 掺杂碳上的过渡金属单原子作为过氧单硫酸盐活化的催化剂:DFT 研究
单原子催化剂在基于过氧单硫酸盐 (PMS) 的高级氧化过程 (AOP) 中表现出色,其中活性氧 (ROS) 的产生对于水中新兴有机污染物的降解至关重要。然而,详细的 PMS 激活机制仍然难以捉摸。密度泛函理论 (DFT) 计算作为一种强大的方法可以克服实验研究的局限性,为催化过程提供分子水平的视角。本研究进行了 DFT 计算,以阐明一系列过渡金属单原子催化剂的电子结构和 PMS 吸附和活化机制。根据 DFT 研究,显着的电子相互作用和负形成能使氮掺杂碳(N@C)载体适用于稳定金属原子(Me)以形成MeN@C催化剂。作为活性位点,单个金属原子通过静电和磁相互作用吸附 PMS 的氧原子,并从 MeN@C 转移电子以激活 PMS。不同的吸附配置和随后的 PMS 活化导致各种 ROS 的产生,包括 SO4 • -自由基、•OH 自由基、单线态氧( 1 O 2 )、高价金属-氧化合物和表面活化的PMS*。由表面激活的 PMS* 介导的电子转移可能在所有 MeN@C/PMS 系统中占主导地位。对于某些系统来说,自由基的产生可能很困难。FeN@C/PMS 和 MnN@C/PMS 容易形成高价金属氧化合物,而CoN@C/PMS、NiN@C/PMS 和 CuN@C/PMS 往往会产生1 O 2 . 该研究结果将为设计和合成用于 AOP 的有效 MeN@C 催化剂以去除水和废水中新兴的有机污染物提供理论基础。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号