当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An Alternative Proposal for the Reaction Mechanism of Light-Dependent Protochlorophyllide Oxidoreductase
ACS Catalysis ( IF 11.3 ) Pub Date : 2022-02-07 , DOI: 10.1021/acscatal.1c05351 Pedro J Silva 1, 2 , Qi Cheng 3, 4
ACS Catalysis ( IF 11.3 ) Pub Date : 2022-02-07 , DOI: 10.1021/acscatal.1c05351 Pedro J Silva 1, 2 , Qi Cheng 3, 4
Affiliation
|
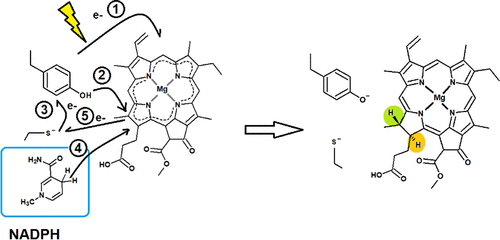
Light-dependent protochlorophyllide oxidoreductase is one of the few known enzymes that require a quantum of light to start their catalytic cycle. Upon excitation, it uses NADPH to reduce the C17–C18 in its substrate (protochlorophyllide) through a complex mechanism that has heretofore eluded precise determination. Isotopic labeling experiments have shown that the hydride-transfer step is very fast, with a small barrier close to 9 kcal mol–1, and is followed by a proton-transfer step, which has been postulated to be the protonation of the product by the strictly conserved Tyr189 residue. Since the structure of the enzyme–substrate complex has not yet been experimentally determined, we first used modeling techniques to discover the actual substrate binding mode. Two possible binding modes were found, both yielding stable binding (as ascertained through molecular dynamics simulations) but only one of which placed the critical C17═C18 bond consistently close to the NADPH pro-S hydrogen and to Tyr189. This binding pose was then used as a starting point for the testing of previous mechanistic proposals using time-dependent density functional theory. The quantum-chemical computations clearly showed that such mechanisms have prohibitively high activation energies. Instead, these computations showed the feasibility of an alternative mechanism initiated by excited-state electron transfer from the key Tyr189 to the substrate. This mechanism appears to agree with the extant experimental data and reinterprets the final protonation step as a proton transfer to the active site itself rather than to the product, aiming at regenerating it for another round of catalysis.
中文翻译:

光依赖性原叶绿素氧化还原酶反应机制的替代方案
光依赖性原叶绿素氧化还原酶是为数不多的需要大量光才能启动其催化循环的已知酶之一。激发后,它使用 NADPH通过迄今为止无法精确确定的复杂机制来减少其底物(原叶绿素)中的 C 17 –C 18 。同位素标记实验表明,氢化物转移步骤非常快,具有接近 9 kcal mol –1的小势垒,然后是质子转移步骤,该步骤被假定为严格保守的 Tyr189 残基对产物的质子化。由于酶-底物复合物的结构尚未通过实验确定,我们首先使用建模技术来发现实际的底物结合模式。发现了两种可能的结合模式,两者都产生稳定的结合(通过分子动力学模拟确定),但只有其中一种将关键的 C17═C18 键始终靠近 NADPH pro - S氢和 Tyr189。然后将该绑定姿势用作使用时间相关密度泛函理论测试先前机械建议的起点。量子化学计算清楚地表明,这种机制具有高得令人望而却步的活化能。相反,这些计算显示了由从关键 Tyr189 到底物的激发态电子转移引发的替代机制的可行性。这种机制似乎与现存的实验数据一致,并将最终的质子化步骤重新解释为质子转移到活性位点本身而不是产物,旨在使其再生以进行另一轮催化。
更新日期:2022-02-07
中文翻译:
光依赖性原叶绿素氧化还原酶反应机制的替代方案
光依赖性原叶绿素氧化还原酶是为数不多的需要大量光才能启动其催化循环的已知酶之一。激发后,它使用 NADPH通过迄今为止无法精确确定的复杂机制来减少其底物(原叶绿素)中的 C 17 –C 18 。同位素标记实验表明,氢化物转移步骤非常快,具有接近 9 kcal mol –1的小势垒,然后是质子转移步骤,该步骤被假定为严格保守的 Tyr189 残基对产物的质子化。由于酶-底物复合物的结构尚未通过实验确定,我们首先使用建模技术来发现实际的底物结合模式。发现了两种可能的结合模式,两者都产生稳定的结合(通过分子动力学模拟确定),但只有其中一种将关键的 C17═C18 键始终靠近 NADPH pro - S氢和 Tyr189。然后将该绑定姿势用作使用时间相关密度泛函理论测试先前机械建议的起点。量子化学计算清楚地表明,这种机制具有高得令人望而却步的活化能。相反,这些计算显示了由从关键 Tyr189 到底物的激发态电子转移引发的替代机制的可行性。这种机制似乎与现存的实验数据一致,并将最终的质子化步骤重新解释为质子转移到活性位点本身而不是产物,旨在使其再生以进行另一轮催化。






























 京公网安备 11010802027423号
京公网安备 11010802027423号