Carbon ( IF 10.5 ) Pub Date : 2022-01-20 , DOI: 10.1016/j.carbon.2022.01.031
Bora Karasulu 1, 2 , Jean-Marc Leyssale 3 , Patrick Rowe 1, 4 , Cedric Weber 5 , Carla de Tomas 1
|
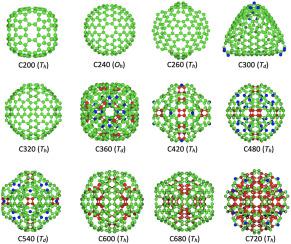
From as small as single carbon dimers up to giant fullerenes or amorphous nanometer-sized particles, the large family of carbon nanoclusters holds a complex structural variability that increases with cluster size. Capturing this variability and predicting stable allotropes remains a challenging modelling task, crucial to advance technological applications of these materials. While small cluster sizes are traditionally investigated with first-principles methods, a comprehensive study spanning larger sizes calls for a computationally efficient alternative. Here, we combine the stochastic ab initio random structure search algorithm (AIRSS) with geometry optimisations based on interatomic potentials to systematically predict the structure of carbon clusters spanning a wide range of sizes. We first test the transferability and predictive capability of seven widely used carbon potentials, including classical and machine-learning potentials. Results are compared against an analogous cluster dataset generated via AIRSS combined with density functional theory optimizations. The best performing potential, GAP-20, is then employed to predict larger clusters in the nanometer scale, overcoming the computational limits of first-principles approaches. Our complete cluster dataset describes the evolution of topological properties with cluster size, capturing the complex variability of the carbon cluster family. As such, the dataset includes ordered and disordered structures, reproducing well-known clusters, like fullerenes, and predicting novel isomers.
中文翻译:
通过结构搜索加速对大型碳簇的预测:机器学习和经典潜力的评估
从小到单个碳二聚体到巨大的富勒烯或无定形纳米级颗粒,碳纳米簇的大家族具有复杂的结构变异性,随着簇大小的增加而增加。捕捉这种可变性并预测稳定的同素异形体仍然是一项具有挑战性的建模任务,对于推进这些材料的技术应用至关重要。虽然传统上使用第一性原理方法研究小集群大小,但跨越较大规模的综合研究需要计算有效的替代方案。在这里,我们结合随机从头算随机结构搜索算法 (AIRSS) 具有基于原子间势的几何优化,以系统地预测跨越各种尺寸的碳簇的结构。我们首先测试了七种广泛使用的碳势的可转移性和预测能力,包括经典和机器学习势。将结果与通过 AIRSS 结合密度泛函理论优化生成的类似集群数据集进行比较。然后使用表现最好的潜力 GAP-20 来预测纳米尺度的更大集群,克服第一性原理方法的计算限制。我们完整的集群数据集描述了拓扑特性随集群大小的演变,捕捉了碳集群家族的复杂可变性。像这样,
































 京公网安备 11010802027423号
京公网安备 11010802027423号