当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Li2S-P2S5 系统的新通用力场
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-01-07 , DOI: 10.1039/d1cp05393k
Shunsuke Ariga 1 , Takahiro Ohkubo 1 , Shingo Urata 2 , Yutaka Imamura 2 , Taketoshi Taniguchi 2
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2022-01-07 , DOI: 10.1039/d1cp05393k
Shunsuke Ariga 1 , Takahiro Ohkubo 1 , Shingo Urata 2 , Yutaka Imamura 2 , Taketoshi Taniguchi 2
Affiliation
|
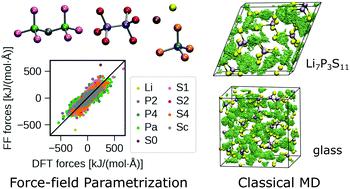
硫代磷酸锂电解液是一种很有前途的全固态电池材料。从头算分子动力学 (AIMD) 模拟已用于研究单晶和玻璃态化合物中的离子传导机制。然而,真实材料(例如,具有晶界和多相玻璃陶瓷的材料)的复杂性导致 AIMD 模拟具有高计算成本。为了克服这一计算限制,我们为锂固态电解质的经典分子动力学 (CMD) 模拟开发了一种新的原子间势。训练数据集由具有代表性的硫化物电解质(β-Li 3 PS 4、γ-Li 3 PS 4、Li 4 P 2 S 6、Li 7 P 3 S 11和 Li 7 PS 6晶体和 70Li 2 S-30P 2 S 5玻璃)。使用 II 类和 Stillinger-Weber 势的函数形式,通过最小化 CMD 和 AIMD 结果之间的原子力、应力和势能差异来优化所有参数。随后的验证表明,优化后的参数可以再现 Li +的动力学以及结晶和玻璃材料的结构。Li 7 P 3 S的离子电导率11晶体大约是等化学计量 70Li 2 S–30P 2 S 5玻璃的 5 倍,表明使用开发的力场的 CMD 模拟准确地再现了AIMD中 Li 7 P 3 S 11中的有效传导路径。开发的力场参数可以在 CMD 框架中模拟复杂材料,包括非晶-晶体界面和多相玻璃陶瓷。

"点击查看英文标题和摘要"
更新日期:2022-01-07
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号