当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electronic Structure of the (Undoped and Fe-Doped) NiOOH O2 Evolution Electrocatalyst
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2016-08-23 00:00:00 , DOI: 10.1021/acs.jpcc.6b06100 José C. Conesa 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2016-08-23 00:00:00 , DOI: 10.1021/acs.jpcc.6b06100 José C. Conesa 1
Affiliation
|
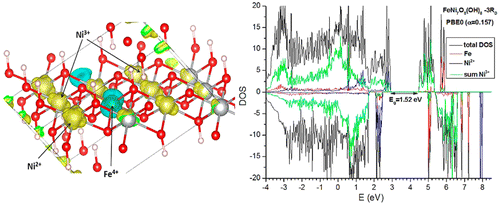
A DFT study was carried out on the atomic and electronic structure of bulk β-NiOOH, with and without substitution of Ni by Fe, examining different stackings and proton distributions in the constituent sheets. The energies of different NiOOH configurations, evaluated at the DFT + U level including dispersion interactions, have very similar values for different stackings and proton distributions, justifying the observed fact that NiOOH is usually obtained in highly disordered form and supporting the assumption that local structures might depend on the synthesis method. Ni ions seem to prefer centrosymmetric coordinations, even if this implies inhomogeneous numbers of OH groups around them; these inhomogeneities can even induce disproportionation of Ni3+ into Ni2+ and Ni4+. The electronic structure, computed with a hybrid DFT functional able to give accurate band gaps for different materials, always shows semiconducting character, with band gaps having widths varying in the 0.8–1.5 eV range and edges formed by Ni 3d states. A metallic character, anticipated by some earlier DFT calculations, is never found. Substitution of Ni by Fe seems to be preferred at sites coordinated with lower numbers of OH groups; this can induce electron transfer from Fe to Ni, that is, formation of Fe4+ and Ni2+, accompanied in some cases by proton jumps across the intersheet space. Some local-level environments, the presence of which might depend on the material synthesis method used, can however stabilize Fe3+, explaining conflicting literature data on the preferred redox state of Fe when included in NiOOH. Overall, the system seems to facilitate electron and proton movement across the material, which can help electrocatalytic O2 evolution processes.
中文翻译:

(未掺杂和铁掺杂)NiOOH O 2析出电催化剂的电子结构
进行了DFT研究,研究了β-NiOOH的原子和电子结构,有无用Fe代替Ni,研究了组成薄片中不同的堆积和质子分布。在DFT + U级评估的包括分散相互作用在内的不同NiOOH构型的能量,对于不同的堆积和质子分布具有非常相似的值,这证明了观察到的事实,即NiOOH通常以高度无序的形式获得,并支持了局部结构可能的假设。取决于合成方法。镍离子似乎更倾向于中心对称配位,即使这暗示着它们周围的OH基团数量不均匀。这些不均匀甚至会导致Ni 3+歧化为Ni 2+和Ni 4+。用混合DFT功能计算的电子结构能够为不同的材料提供准确的带隙,该电子结构始终显示出半导体特性,其带隙的宽度在0.8-1.5 eV范围内变化,并且边缘由Ni 3d态形成。从未发现过一些DFT计算所预期的金属特征。在与较少数量的OH基团配位的位点似乎更倾向于用Fe取代Ni。这会引起电子从Fe转移到Ni,即形成Fe 4+和Ni 2+,在某些情况下还伴随着质子跃过层间空间。但是,某些局部环境的存在可能取决于所使用的材料合成方法,但它们可以稳定Fe 3+解释了有关NiOOH中优选的Fe氧化还原态的文献资料。总的来说,该系统似乎促进了电子和质子在整个材料上的运动,这可以帮助电催化O 2的释放过程。
更新日期:2016-08-23
中文翻译:
(未掺杂和铁掺杂)NiOOH O 2析出电催化剂的电子结构
进行了DFT研究,研究了β-NiOOH的原子和电子结构,有无用Fe代替Ni,研究了组成薄片中不同的堆积和质子分布。在DFT + U级评估的包括分散相互作用在内的不同NiOOH构型的能量,对于不同的堆积和质子分布具有非常相似的值,这证明了观察到的事实,即NiOOH通常以高度无序的形式获得,并支持了局部结构可能的假设。取决于合成方法。镍离子似乎更倾向于中心对称配位,即使这暗示着它们周围的OH基团数量不均匀。这些不均匀甚至会导致Ni 3+歧化为Ni 2+和Ni 4+。用混合DFT功能计算的电子结构能够为不同的材料提供准确的带隙,该电子结构始终显示出半导体特性,其带隙的宽度在0.8-1.5 eV范围内变化,并且边缘由Ni 3d态形成。从未发现过一些DFT计算所预期的金属特征。在与较少数量的OH基团配位的位点似乎更倾向于用Fe取代Ni。这会引起电子从Fe转移到Ni,即形成Fe 4+和Ni 2+,在某些情况下还伴随着质子跃过层间空间。但是,某些局部环境的存在可能取决于所使用的材料合成方法,但它们可以稳定Fe 3+解释了有关NiOOH中优选的Fe氧化还原态的文献资料。总的来说,该系统似乎促进了电子和质子在整个材料上的运动,这可以帮助电催化O 2的释放过程。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号