当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
机器学习化学反应的活化能
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2021-12-30 , DOI: 10.1002/wcms.1593 Toby Lewis‐Atwell 1 , Piers A. Townsend 2 , Matthew N. Grayson 2
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2021-12-30 , DOI: 10.1002/wcms.1593 Toby Lewis‐Atwell 1 , Piers A. Townsend 2 , Matthew N. Grayson 2
Affiliation
|
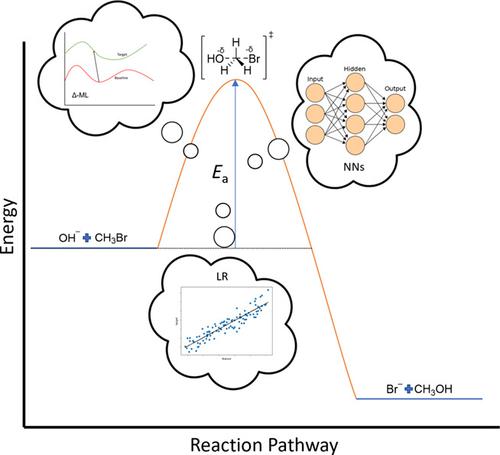
将机器学习 (ML) 应用于预测反应激活障碍是这些算法的一个新的令人兴奋的领域。这里涉及的工作特别是那些训练 ML 以预测均相化学反应的活化能的工作,其中活化能由反应物之间的能量差和反应的过渡态给出。特别关注应用 ML 直接预测反应活化能的工作、这些研究中可能发现的局限性,以及对 ML 模型的不同类型化学特征进行比较的地方。还探索了使用高斯过程回归 ML 模型能够获得高预测准确度但数据集减少的模型。在这些研究中,模拟活化势垒的化学反应包括那些涉及有机小分子、芳香环和有机金属催化剂的化学反应。还提供了对化学中使用的一些最流行的 ML 模型类型的简要说明,作为不熟悉的初学者的指南。

"点击查看英文标题和摘要"
更新日期:2021-12-30
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号