Journal of Structural Chemistry ( IF 1.2 ) Pub Date : 2021-12-15 , DOI: 10.1134/s0022476621110111 A. Saeed 1 , G. Shabir 1 , P. A. Channar 1 , U. Flörke 2 , T. Hökelek 3 , M. F. Erben 4
|
Abstract
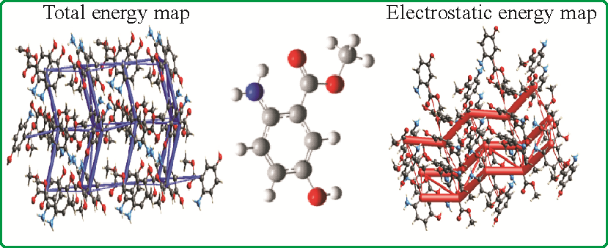
The title compound with the molecular formula C8H9NO3 is synthesized by refluxing 2-amino-5-hydroxybenzoic acid in methanol. The molecular structure of the compound is determined by single crystal X-ray diffraction. Methyl 2-amino-5-hydroxybenzoate crystallizes in the orthorhombic space group P212121 with a = 4.973(2) Å, b = 10.923(5) Å, c = 14.074(6) Å, Z = 4 and V = 764.4(6) Å3. DFT is used to compute HOMO–LUMO energy levels, to predict the reactivity of substituents (NH2 and OH), and to determine the nucleophilic character of these two groups. The orientation and nature of substituents on benzene favors the formation of a stable six-membered ring via hydrogen bonding which plays a key role in the properties of the investigated compound. The natural bond orbital (NBO) population analysis demonstrates that the hyperconjugative effect between the donor lone pairs located on the carbonyl oxygen atom and the N–H group, via the lp O → σ*(N–H) 1,6-remote interaction, is responsible for the preferred conformation. The molecular electrostatic potential (MEP) surface shows the electrical neutrality in the molecule. To get an insight to the intermolecular interactions in the crystal a Hirshfeld surface analysis is also carried out.
中文翻译:
2-氨基-5-羟基苯甲酸甲酯的计算研究、HIRSHFELD 表面分析、相互作用能计算和能量框架晶体结构
摘要
通过在甲醇中回流2-氨基-5-羟基苯甲酸合成分子式为C 8 H 9 NO 3的标题化合物。该化合物的分子结构由单晶X射线衍射确定。正交空间群中的2-氨基-5-羟基苯甲酸甲酯结晶P 2 1 2 1 2 1与一个 = 4.973(2)埃,b = 10.923(5),C ^ = 14.074(6)A,Ž = 4和V = 764.4(6) Å 3。DFT 用于计算 HOMO-LUMO 能级,以预测取代基 (NH 2和 OH),并确定这两个基团的亲核特性。苯上取代基的取向和性质有利于通过氢键形成稳定的六元环,这在所研究化合物的性质中起着关键作用。自然键轨道 (NBO) 布居分析表明,位于羰基氧原子上的供体孤对与 N–H 基团之间的超共轭效应,通过 l p O → σ * (N–H) 1,6-remote相互作用,负责首选构象。分子静电势 (MEP) 表面显示分子中的电中性。为了深入了解晶体中的分子间相互作用,还进行了 Hirshfeld 表面分析。










































 京公网安备 11010802027423号
京公网安备 11010802027423号