Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Molecular Dynamics Simulations of the Thermal Decomposition of 3,4-Bis(3-nitrofurazan-4-yl)furoxan
ACS Omega ( IF 3.7 ) Pub Date : 2021-12-05 , DOI: 10.1021/acsomega.1c04166 Yang Li 1 , Yucun Liu 1 , Junming Yuan 1 , Yiming Luo 2 , Qiuli Jiang 2 , Fanfan Wang 1, 3 , Jingwei Meng 1
ACS Omega ( IF 3.7 ) Pub Date : 2021-12-05 , DOI: 10.1021/acsomega.1c04166 Yang Li 1 , Yucun Liu 1 , Junming Yuan 1 , Yiming Luo 2 , Qiuli Jiang 2 , Fanfan Wang 1, 3 , Jingwei Meng 1
Affiliation
|
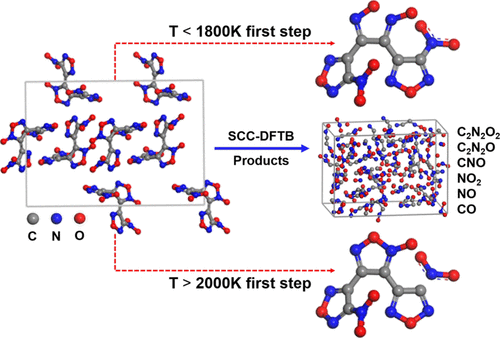
When stimulated, for example, by a high temperature, the physical and chemical properties of energetic materials (EMs) may change, and, in turn, their overall performance is affected. Therefore, thermal stability is crucial for EMs, especially the thermal dynamic behavior. In the past decade, significant efforts have been made to study the thermal dynamic behavior of 3,4-bis(3-nitrofurazan-4-yl)furoxan (DNTF), one of the new high-energy-density materials (HEDMs). However, the thermal decomposition mechanism of DNTF is still not specific or comprehensive. In this study, the self-consistent-charge density-functional tight-binding method was combined with molecular dynamics (MD) simulations to reveal the differences in the thermal decomposition of DNTF under four heating conditions. The O–N (O) bond would fracture first during DNTF initial thermal decomposition at medium and low temperatures, thus triggering the cracking of the whole structure. At 2000 and 2500 K, NO2 loss on outer ring I is the fastest initial thermal decomposition pathway, and it determines that the decomposition mechanism is different from that of a medium-low temperature. NO2 is found to be the most active intermediate product; large molecular fragments, such as C2N2O, are found for the first time. Hopefully, these results could provide some insights into the decomposition mechanism of new HEDMs.
中文翻译:

3,4-双(3-硝基呋喃-4-基)呋喃的热分解的分子动力学模拟
例如,当受到高温刺激时,含能材料 (EM) 的物理和化学性质可能会发生变化,进而影响其整体性能。因此,热稳定性对 EM 至关重要,尤其是热动态行为。在过去的十年中,人们为研究 3,4-双(3-硝基呋喃-4-基)呋喃 (DNTF) 的热力学行为做出了重大努力,该材料是一种新型高能量密度材料 (HEDM)。然而,DNTF的热分解机理仍不具体或不全面。在这项研究中,自洽电荷密度泛函紧束缚方法与分子动力学(MD)模拟相结合,揭示了 DNTF 在四种加热条件下热分解的差异。O-N(O)键在中低温下DNTF初始热分解过程中首先断裂,从而引发整个结构的开裂。在 2000 和 2500 K 时,NO外环Ⅰ上的2损失是最快的初始热分解途径,它决定了分解机理与中低温不同。发现NO 2是最活跃的中间产物;首次发现大分子碎片,如 C 2 N 2 O。希望这些结果可以为新 HEDM 的分解机制提供一些见解。
更新日期:2021-12-14
中文翻译:
3,4-双(3-硝基呋喃-4-基)呋喃的热分解的分子动力学模拟
例如,当受到高温刺激时,含能材料 (EM) 的物理和化学性质可能会发生变化,进而影响其整体性能。因此,热稳定性对 EM 至关重要,尤其是热动态行为。在过去的十年中,人们为研究 3,4-双(3-硝基呋喃-4-基)呋喃 (DNTF) 的热力学行为做出了重大努力,该材料是一种新型高能量密度材料 (HEDM)。然而,DNTF的热分解机理仍不具体或不全面。在这项研究中,自洽电荷密度泛函紧束缚方法与分子动力学(MD)模拟相结合,揭示了 DNTF 在四种加热条件下热分解的差异。O-N(O)键在中低温下DNTF初始热分解过程中首先断裂,从而引发整个结构的开裂。在 2000 和 2500 K 时,NO外环Ⅰ上的2损失是最快的初始热分解途径,它决定了分解机理与中低温不同。发现NO 2是最活跃的中间产物;首次发现大分子碎片,如 C 2 N 2 O。希望这些结果可以为新 HEDM 的分解机制提供一些见解。






































 京公网安备 11010802027423号
京公网安备 11010802027423号