当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab Initio Study of the C–O Bond Dissociation in CO2 Reduction by Redox and Carboxyl Routes on 3d Transition Metal Systems
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2021-11-24 , DOI: 10.1021/acs.jpcc.1c05468 Vivianne K. Ocampo-Restrepo 1 , Lucas G. Verga 1 , Juarez L. F. Da Silva 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2021-11-24 , DOI: 10.1021/acs.jpcc.1c05468 Vivianne K. Ocampo-Restrepo 1 , Lucas G. Verga 1 , Juarez L. F. Da Silva 1
Affiliation
|
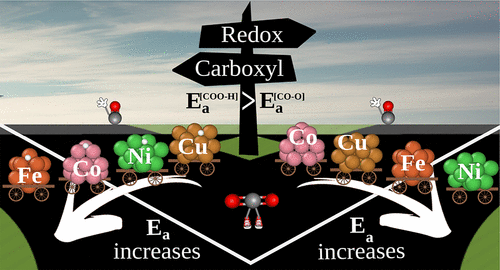
The C–O bond dissociation of the CO2 molecule via the reverse water gas shift reaction is crucial for several reactions used as renewable alternatives for fuel synthesis. However, our atomistic understanding of this process on transition metal (TM) clusters, where quantum-size effects might play a significant role, is far from complete. Here, we addressed the C–O bond dissociation by redox and carboxyl routes on 13-atom TM (Fe, Co, Ni, Cu) clusters using density functional theory calculations and the climbing image nudged elastic band algorithm. From the potential energy profiles, we found that CO2 is prone to dissociate into adsorbed CO via the redox route with lower activation energies, Ea’s, than the carboxyl route on all studied TM13 systems. Our results suggested that the smaller activation barrier found on the Co13 cluster is due to the stronger adsorption exhibited for both CO2 and O. By increasing the d-state occupation (from Fe to Cu), the Ea differences between CO2 dissociation and COOH formation decrease. We associated this behavior with a decrease in the (CO2 + H) adsorption energy from Fe13 to Cu13 that facilitates the COO–H bond formation and H–TM bond cleavage, i.e., favoring the carboxyl route. Also, our analyses indicate that the adsorption energies of the CO2 and trans-COOH species are the best descriptors for the C–O bond dissociation via the redox and carboxyl routes, respectively.
中文翻译:

在 3d 过渡金属系统上通过氧化还原和羧基途径还原 CO2 中 C-O 键离解的 Ab Initio 研究
CO 2分子通过反向水煤气变换反应的 C-O 键解离对于用作燃料合成的可再生替代品的几种反应至关重要。然而,我们对过渡金属 (TM) 簇(其中量子尺寸效应可能起重要作用)的这一过程的原子理解还远未完成。在这里,我们使用密度泛函理论计算和爬升图像轻推弹力带算法,通过氧化还原和羧基路线在 13 原子 TM(Fe、Co、Ni、Cu)簇上解决了 C-O 键解离。从势能分布中,我们发现 CO 2易于通过氧化还原途径分解成吸附的 CO,其活化能E a比所有研究的 TM 上的羧基途径都低13个系统。我们的结果表明,在 Co 13簇上发现的较小活化势垒是由于对 CO 2和 O表现出更强的吸附。通过增加 d 态占据(从 Fe 到 Cu),CO 2解离之间的E a差异和 COOH 形成减少。我们将此行为与从 Fe 13到 Cu 13的 (CO 2 + H) 吸附能的降低相关联,这促进了 COO-H 键的形成和 H-TM 键的断裂,即有利于羧基路线。此外,我们的分析表明,CO 2和反式的吸附能-COOH 物质分别是通过氧化还原和羧基途径进行 C-O 键解离的最佳描述词。
更新日期:2021-12-09
中文翻译:
在 3d 过渡金属系统上通过氧化还原和羧基途径还原 CO2 中 C-O 键离解的 Ab Initio 研究
CO 2分子通过反向水煤气变换反应的 C-O 键解离对于用作燃料合成的可再生替代品的几种反应至关重要。然而,我们对过渡金属 (TM) 簇(其中量子尺寸效应可能起重要作用)的这一过程的原子理解还远未完成。在这里,我们使用密度泛函理论计算和爬升图像轻推弹力带算法,通过氧化还原和羧基路线在 13 原子 TM(Fe、Co、Ni、Cu)簇上解决了 C-O 键解离。从势能分布中,我们发现 CO 2易于通过氧化还原途径分解成吸附的 CO,其活化能E a比所有研究的 TM 上的羧基途径都低13个系统。我们的结果表明,在 Co 13簇上发现的较小活化势垒是由于对 CO 2和 O表现出更强的吸附。通过增加 d 态占据(从 Fe 到 Cu),CO 2解离之间的E a差异和 COOH 形成减少。我们将此行为与从 Fe 13到 Cu 13的 (CO 2 + H) 吸附能的降低相关联,这促进了 COO-H 键的形成和 H-TM 键的断裂,即有利于羧基路线。此外,我们的分析表明,CO 2和反式的吸附能-COOH 物质分别是通过氧化还原和羧基途径进行 C-O 键解离的最佳描述词。






























 京公网安备 11010802027423号
京公网安备 11010802027423号