当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electrocatalytic Reduction of CO2 to CO over Ag(110) and Cu(211) Modeled by Grand-Canonical Density Functional Theory
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2021-10-21 , DOI: 10.1021/acs.jpcc.1c07484 Yousef A. Alsunni 1, 2 , Abdulaziz W. Alherz 2 , Charles B. Musgrave 2, 3, 4
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2021-10-21 , DOI: 10.1021/acs.jpcc.1c07484 Yousef A. Alsunni 1, 2 , Abdulaziz W. Alherz 2 , Charles B. Musgrave 2, 3, 4
Affiliation
|
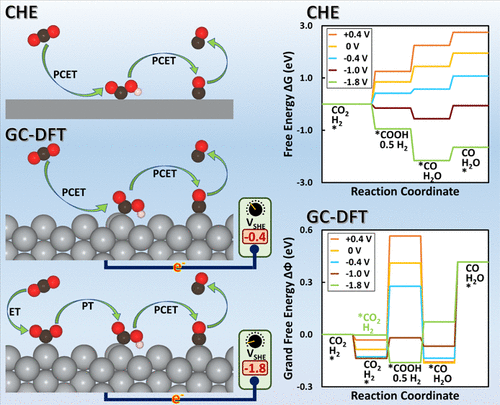
We report the results of modeling CO2 reduction (CO2R) to CO over Ag(110) and Cu(211) surfaces at different applied potentials using grand-canonical density functional theory (GC-DFT), a method specifically designed to accurately model electrochemical systems. In addition to demonstrating GC-DFT’s ability to accurately model electrochemical processes, we also compare it with the computational hydrogen electrode (CHE) approach. GC-DFT predicts that the geometries of these reacting systems strongly depend on the applied potential, and the Helmholtz free energies vary nonlinearly with the applied potential, which contradicts a central assumption of the CHE approach. The CHE approach neglects the change in the number of electrons on the electrode surface at different applied potentials, which reduces its accuracy as the potential changes from the potential of zero charge. Our results further demonstrate that the grand free energies of the reaction intermediates not only depend on the value of the applied potential but also on the metal surface type, adsorption site, and adsorbate. GC-DFT’s ability to predict the effect of the applied potential on adsorbate geometry enables it to evaluate different possible reaction mechanisms at different applied potentials. For instance, GC-DFT predicts that the first step of CO2R likely switches from proton-coupled electron transfer to sequential electron transfer and then proton transfer at more reducing potentials, a result that cannot be determined by the CHE because it assumes that all electron transfers are coupled to proton transfers and neglects the effect of the applied potential on the adsorbate geometry.
中文翻译:

通过正则密度泛函理论建模的 Ag(110) 和 Cu(211) 上的 CO2 电催化还原为 CO
我们报告了模拟 CO 2还原(CO 2R) 在不同外加电位下在 Ag(110) 和 Cu(211) 表面上转化为 CO,使用大正则密度泛函理论 (GC-DFT),这是一种专门设计用于准确模拟电化学系统的方法。除了展示 GC-DFT 准确模拟电化学过程的能力外,我们还将其与计算氢电极 (CHE) 方法进行了比较。GC-DFT 预测这些反应系统的几何形状在很大程度上取决于施加的电位,并且亥姆霍兹自由能随施加的电位非线性变化,这与 CHE 方法的中心假设相矛盾。CHE 方法忽略了在不同外加电位下电极表面电子数的变化,随着电位从零电荷电位变化而降低了其准确性。我们的结果进一步表明,反应中间体的大自由能不仅取决于施加的电位值,还取决于金属表面类型、吸附位点和吸附质。GC-DFT 能够预测施加电位对吸附物几何形状的影响,使其能够评估不同施加电位下不同可能的反应机制。例如,GC-DFT 预测 CO 的第一步2 R 可能从质子耦合电子转移切换到顺序电子转移,然后在更多还原电位下进行质子转移,CHE 无法确定这一结果,因为它假设所有电子转移都与质子转移耦合并忽略了在吸附物几何形状上施加电位。
更新日期:2021-11-04
中文翻译:
通过正则密度泛函理论建模的 Ag(110) 和 Cu(211) 上的 CO2 电催化还原为 CO
我们报告了模拟 CO 2还原(CO 2R) 在不同外加电位下在 Ag(110) 和 Cu(211) 表面上转化为 CO,使用大正则密度泛函理论 (GC-DFT),这是一种专门设计用于准确模拟电化学系统的方法。除了展示 GC-DFT 准确模拟电化学过程的能力外,我们还将其与计算氢电极 (CHE) 方法进行了比较。GC-DFT 预测这些反应系统的几何形状在很大程度上取决于施加的电位,并且亥姆霍兹自由能随施加的电位非线性变化,这与 CHE 方法的中心假设相矛盾。CHE 方法忽略了在不同外加电位下电极表面电子数的变化,随着电位从零电荷电位变化而降低了其准确性。我们的结果进一步表明,反应中间体的大自由能不仅取决于施加的电位值,还取决于金属表面类型、吸附位点和吸附质。GC-DFT 能够预测施加电位对吸附物几何形状的影响,使其能够评估不同施加电位下不同可能的反应机制。例如,GC-DFT 预测 CO 的第一步2 R 可能从质子耦合电子转移切换到顺序电子转移,然后在更多还原电位下进行质子转移,CHE 无法确定这一结果,因为它假设所有电子转移都与质子转移耦合并忽略了在吸附物几何形状上施加电位。
































 京公网安备 11010802027423号
京公网安备 11010802027423号