Letters in Drug Design & Discovery ( IF 1.2 ) Pub Date : 2021-07-31 , DOI: 10.2174/1570180818666210114115030 W.U. Lu-Yang 1 , M.A. Yang-Min 1 , L.E.I. Shan 1 , Wang Tian-Hao 1 , Feng Yi 1
|
Background: Malaria is one of the most important infectious diseases in the world. The most severe form of malaria in humans is caused by Plasmodium falciparum. Malaria is a worldwide health problem, with 214 million new cases in 2015 and 438,000 deaths, most of which are in Africa. Therefore, there is an urgent need for novel, low-toxic, more specific inhibitors to find new antimalarial agents. A promising target for antimalarial drug design is falcipain-2, a cysteine protease from P. falciparum that has received considerable attention due to its key role in the life cycle of the parasite.
Materials and Methods: Three-dimensional quantitative structure-activity relationship (3D-QSAR) models of 39 peptidyl vinyl sulfone cysteine protease inhibitors was constructed using Topomer CoMFA. Topomer Search was employed to virtually screen lead-like compounds in the ZINC database. Molecular docking was employed to further explore the binding requirements between the ligands and the receptor protein which included several hydrogen bonds between peptidyl vinyl sulfone cysteine protease inhibitors and active site residues.
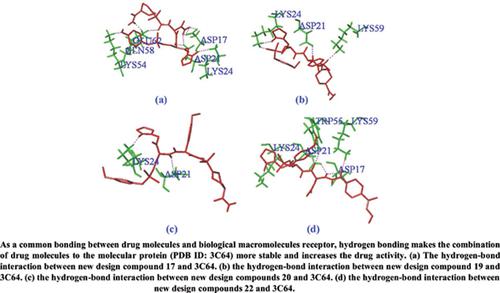
Results: The non-cross correlation coefficient (r2), the interaction validation coefficient (q2) and the external validation (r2 pred) were 0.902, 0.685 and 0.763, respectively. The results showed that the model not only had good estimation stability but also good prediction capability. 22 new molecules were obtained, whose predicted activity is higher than the template molecules. The results showed that the Topomer Search technology can be effectively applied to screen and design new peptidyl vinyl sulfone cysteine protease inhibitors. Molecular docking showed extensive interactions between peptidyl vinyl sulfone cysteine protease inhibitors and residues of LYS24, ASP21, LYS59, and ASP17 in the active site.
Conclusion: 39 peptidyl vinyl sulfone cysteine protease inhibitors were used in the 3D-QSAR study. Topomer CoMFA 3D-QSAR method was used to build the model, and the model was well predicted and statistically validated. The design of potent new inhibitors of cysteine protease can get useful insights from these results.
中文翻译:
使用拓扑异构体 CoMFA 和分子对接对肽基乙烯基砜半胱氨酸蛋白酶抑制剂进行 QSAR 研究
背景:疟疾是世界上最重要的传染病之一。人类最严重的疟疾是由恶性疟原虫引起的。疟疾是一个全球性的健康问题,2015 年有 2.14 亿新病例和 438,000 人死亡,其中大部分在非洲。因此,迫切需要新的、低毒的、更特异的抑制剂来寻找新的抗疟药。抗疟药物设计的一个有希望的目标是 falcipain-2,这是一种来自恶性疟原虫的半胱氨酸蛋白酶,由于其在寄生虫生命周期中的关键作用而受到相当多的关注。
材料和方法:使用 Topomer CoMFA 构建了 39 种肽基乙烯基砜半胱氨酸蛋白酶抑制剂的三维定量构效关系 (3D-QSAR) 模型。Topomer Search 用于在 ZINC 数据库中虚拟筛选铅样化合物。分子对接被用来进一步探索配体和受体蛋白之间的结合要求,包括肽基乙烯基砜半胱氨酸蛋白酶抑制剂和活性位点残基之间的几个氢键。
结果:非互相关系数(r 2)、交互验证系数(q 2)和外部验证(r 2 pred)分别为0.902、0.685和0.763。结果表明,该模型不仅具有较好的估计稳定性,而且具有较好的预测能力。获得了22个新分子,其预测活性高于模板分子。结果表明,Topomer Search技术可有效应用于筛选和设计新型肽基乙烯基砜半胱氨酸蛋白酶抑制剂。分子对接显示肽基乙烯基砜半胱氨酸蛋白酶抑制剂与活性位点中 LYS24、ASP21、LYS59 和 ASP17 的残基之间存在广泛的相互作用。
结论:39 种肽基乙烯基砜半胱氨酸蛋白酶抑制剂用于 3D-QSAR 研究。采用Topomer CoMFA 3D-QSAR方法建立模型,模型得到较好的预测和统计验证。有效的新半胱氨酸蛋白酶抑制剂的设计可以从这些结果中获得有用的见解。
































 京公网安备 11010802027423号
京公网安备 11010802027423号