当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Origin of the N-coordinated single-atom Ni sites in heterogeneous electrocatalysts for CO2 reduction reaction
Chemical Science ( IF 7.6 ) Pub Date : 2021-10-07 , DOI: 10.1039/d1sc04094d Yu Wang 1 , Liming You 1 , Kun Zhou 1, 2
Chemical Science ( IF 7.6 ) Pub Date : 2021-10-07 , DOI: 10.1039/d1sc04094d Yu Wang 1 , Liming You 1 , Kun Zhou 1, 2
Affiliation
|
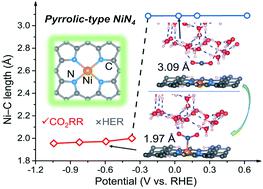
Heterogeneous Ni–N–C single-atom catalysts (SACs) have attracted great research interest regarding their capability in facilitating the CO2 reduction reaction (CO2RR), with CO accounting for the major product. However, the fundamental nature of their active Ni sites remains controversial, since the typically proposed pyridinic-type Ni configurations are inactive, display low selectivity, and/or possess an unfavorable formation energy. Herein, we present a constant-potential first-principles and microkinetic model to study the CO2RR at a solid–water interface, which shows that the electrode potential is crucial for governing CO2 activation. A formation energy analysis on several NiNxC4−x (x = 1–4) moieties indicates that the predominant Ni moieties of Ni–N–C SACs are expected to have a formula of NiN4. After determining the potential-dependent thermodynamic and kinetic energy of these Ni moieties, we discover that the energetically favorable pyrrolic-type NiN4 moiety displays high activity for facilitating the selective CO2RR over the competing H2 evolution. Moreover, model polarization curves and Tafel analysis results exhibit reasonable agreement with existing experimental data. This work highlights the intrinsic tetrapyrrolic coordination of Ni for facilitating the CO2RR and offers practical guidance for the rational improvement of SACs, and this model can be expanded to explore mechanisms of other electrocatalysis in aqueous solutions.
中文翻译:

CO2还原反应多相电催化剂中N配位单原子Ni位点的起源
非均相Ni-N-C单原子催化剂(SAC)因其促进CO 2还原反应(CO 2 RR)的能力而引起了人们极大的研究兴趣,其中CO是主要产物。然而,其活性镍位点的基本性质仍然存在争议,因为通常提出的吡啶型镍构型是不活泼的,表现出低选择性,和/或具有不利的形成能。在此,我们提出了恒电位第一原理和微动力学模型来研究固水界面上的CO 2 RR,这表明电极电位对于控制CO 2活化至关重要。对几个 NiN x C 4− x ( x = 1–4) 部分的形成能分析表明 Ni-N-C SAC 的主要 Ni 部分预计具有 NiN 4的分子式。在确定这些Ni部分的电势依赖性热力学和动能后,我们发现能量上有利的吡咯型NiN 4部分表现出高活性,有利于选择性CO 2 RR而不是竞争性H 2析出。此外,模型极化曲线和塔菲尔分析结果与现有实验数据表现出合理的一致性。这项工作突出了Ni内在的四吡咯配位促进CO 2 RR的作用,为SAC的合理改进提供了实践指导,并且该模型可以扩展到探索水溶液中其他电催化的机制。
更新日期:2021-10-13
中文翻译:
CO2还原反应多相电催化剂中N配位单原子Ni位点的起源
非均相Ni-N-C单原子催化剂(SAC)因其促进CO 2还原反应(CO 2 RR)的能力而引起了人们极大的研究兴趣,其中CO是主要产物。然而,其活性镍位点的基本性质仍然存在争议,因为通常提出的吡啶型镍构型是不活泼的,表现出低选择性,和/或具有不利的形成能。在此,我们提出了恒电位第一原理和微动力学模型来研究固水界面上的CO 2 RR,这表明电极电位对于控制CO 2活化至关重要。对几个 NiN x C 4− x ( x = 1–4) 部分的形成能分析表明 Ni-N-C SAC 的主要 Ni 部分预计具有 NiN 4的分子式。在确定这些Ni部分的电势依赖性热力学和动能后,我们发现能量上有利的吡咯型NiN 4部分表现出高活性,有利于选择性CO 2 RR而不是竞争性H 2析出。此外,模型极化曲线和塔菲尔分析结果与现有实验数据表现出合理的一致性。这项工作突出了Ni内在的四吡咯配位促进CO 2 RR的作用,为SAC的合理改进提供了实践指导,并且该模型可以扩展到探索水溶液中其他电催化的机制。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号