当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
使用优化的准正则高斯基和搭配方法计算分子光谱
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-10-12 , DOI: 10.1021/acs.jctc.1c00805
Shane W Flynn 1 , Vladimir A Mandelshtam 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-10-12 , DOI: 10.1021/acs.jctc.1c00805
Shane W Flynn 1 , Vladimir A Mandelshtam 1
Affiliation
|
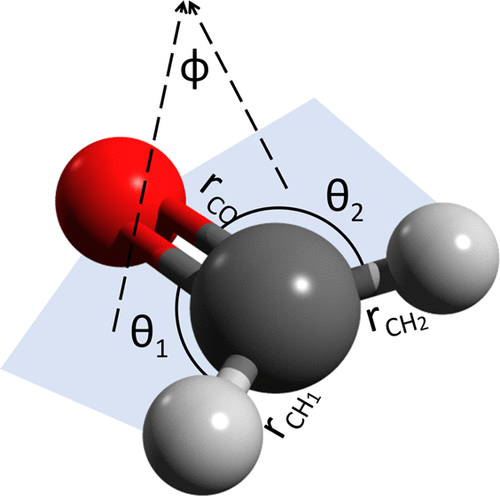
我们重新审视 Manzhos 和 Carrington 的搭配方法 [ J. Chem. 物理。, 2016 , 145 ,224110 ] 其中分布式局部(例如,高斯)基用于建立广义特征值问题,以计算分子振动哈密顿量的特征能量和特征函数。虽然由此产生的线性代数问题涉及全矩阵,但该方法提供了许多重要的优点,即(i)它在概念上和数值上都非常简单,(ii)它可以使用任何一组内部分子坐标来公式化,(iii) ) 它在基的选择方面是灵活的,(iv) 不需要计算积分,并且 (v) 它有可能通过优化基函数的位置和形状来显着减小基大小。在本文中,我们使用最近引入的、这里进一步改进的准规则网格 (QRG) 来探索该方法的后一方面。

"点击查看英文标题和摘要"
更新日期:2021-11-09
"点击查看英文标题和摘要"







































 京公网安备 11010802027423号
京公网安备 11010802027423号