当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
如何在蛋白质力场中达到构象平衡以进行分子动力学模拟?
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2021-10-05 , DOI: 10.1002/wcms.1578
Wei Kang 1 , Fan Jiang 2 , Yun‐Dong Wu 1, 2, 3
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2021-10-05 , DOI: 10.1002/wcms.1578
Wei Kang 1 , Fan Jiang 2 , Yun‐Dong Wu 1, 2, 3
Affiliation
|
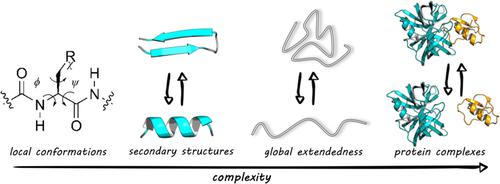
分子动力学 (MD) 模拟是探索蛋白质构象能量景观的有力工具,MD 结果的可靠性关键取决于潜在的力场 (FF)。能够在多个层次上产生不同构象的平衡分布的准确 FF 一直是一个长期追求的目标。为此,已经进行了几十年的共同努力来解决 FF 缺陷,表现为不同水平的构象偏差(局部构象、二级结构和多肽链的全局延伸)。我们首先介绍主要的 FF 偏差,然后回顾分别解决它们的策略。具体来说,扭转参数优化的非残基特异性和残基特异性策略均已应用于实现局部构象和二级结构平衡。使用特定于残基的扭转参数可以获得显着的改进,特别是在考虑显式二面耦合时。此外,蛋白质-蛋白质和蛋白质-水相互作用之间的额外平衡已通过多种方式进行优化,以重现多肽链的全局延伸性,特别是对于未折叠或无序的蛋白质。这篇综述旨在总结过去最有价值的经验和教训,我们希望这可以促进经典 FF 和更复杂的模型(如可极化 FF)的进一步改进。使用特定于残基的扭转参数可以获得显着的改进,特别是在考虑显式二面耦合时。此外,蛋白质-蛋白质和蛋白质-水相互作用之间的额外平衡已通过多种方式进行优化,以重现多肽链的全局延伸性,特别是对于未折叠或无序的蛋白质。这篇综述旨在总结过去最有价值的经验和教训,我们希望这可以促进经典 FF 和更复杂的模型(如可极化 FF)的进一步改进。使用特定于残基的扭转参数可以获得显着的改进,特别是在考虑显式二面耦合时。此外,蛋白质-蛋白质和蛋白质-水相互作用之间的额外平衡已通过多种方式进行优化,以重现多肽链的全局延伸性,特别是对于未折叠或无序的蛋白质。这篇综述旨在总结过去最有价值的经验和教训,我们希望这可以促进经典 FF 和更复杂的模型(如可极化 FF)的进一步改进。特别是对于未折叠或无序的蛋白质。这篇综述旨在总结过去最有价值的经验和教训,我们希望这可以促进经典 FF 和更复杂的模型(如可极化 FF)的进一步改进。特别是对于未折叠或无序的蛋白质。这篇综述旨在总结过去最有价值的经验和教训,我们希望这可以促进经典 FF 和更复杂的模型(如可极化 FF)的进一步改进。

"点击查看英文标题和摘要"
更新日期:2021-10-05
"点击查看英文标题和摘要"



































 京公网安备 11010802027423号
京公网安备 11010802027423号