Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-08-25 , DOI: 10.1016/j.molstruc.2021.131367 Qingmei Wu 1, 2 , Zhaopeng Zheng 3 , Wenjun Ye 1, 2 , Qian Guo 1, 2 , Tianhui Liao 1, 2 , Di Yang 1, 2 , Chunshen Zhao 1, 2 , Weike Liao 4 , Huifang Chai 5 , Zhixu Zhou 1, 2, 6
|
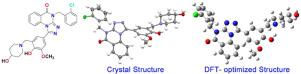
In current work, we have firstly synthesized 4-(2-chlorobenzyl)-1-(4‑hydroxy-3- ((4-hydroxypiperidin-1-yl)methyl)-5-methoxyphenyl)-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one (1). The structural properties of 1 were explored using spectroscopy (1H NMR, 13C NMR, MS and FT-IR) and X-ray crystallography method. The single-crystal structure confirmed by X-ray diffraction was consistent with the molecular structure optimized by density functional theory (DFT) calculation at B3LYP/6–311 G (2d, p) level of theory. The geometrical parameters, molecular electrostatic potential (MEP) and frontier molecular orbital (FMO) analysis were performed by DFT using the B3LYP/6–311 G (2d, p) method. Molecular docking may suggest a favorable interaction between 1 and SHP2 protein. The molecular dynamics (MD) simulation results shown that there are hydrogen bonds, electrostatic interactions and Pi interactions between compound 1 and SHP2 proteins. The inhibitory activity of 1 on SHP2 protein at 10 μM is better than the reference compound (SHP244).
中文翻译:
4-(2-氯苄基)-1-(4-羟基-3-((4-羟基哌啶-1-基)甲基-5-甲氧基苯基)-[的合成、晶体和分子结构、振动光谱、DFT和分子对接1,2,4] 三唑并 [4,3-a] quinazolin-5(4H)-one
在目前的工作中,我们首先合成了4-(2-氯苄基)-1-(4-羟基-3-((4-羟基哌啶-1-基)甲基)-5-甲氧基苯基)-[1,2,4]三唑并[4,3 - a ]quinazolin-5(4 H )-one ( 1 )。的结构性质1使用光谱(进行了探索1 H NMR,13C NMR、MS 和 FT-IR) 和 X 射线晶体学方法。通过 X 射线衍射证实的单晶结构与在 B3LYP/6-311 G (2d, p) 理论水平下通过密度泛函理论 (DFT) 计算优化的分子结构一致。几何参数、分子静电势 (MEP) 和前沿分子轨道 (FMO) 分析通过 DFT 使用 B3LYP/6-311 G (2d, p) 方法进行。分子对接可能表明1和 SHP2 蛋白之间存在有利的相互作用。分子动力学(MD)模拟结果表明,化合物1与SHP2蛋白之间存在氢键、静电相互作用和Pi相互作用。1的抑制活性10 μM 对 SHP2 蛋白的影响优于参考化合物 ( SHP244 )。







































 京公网安备 11010802027423号
京公网安备 11010802027423号