Redox Biology ( IF 10.7 ) Pub Date : 2021-08-18 , DOI: 10.1016/j.redox.2021.102108 Tzong-Shyuan Lee, Tse-Min Lu, Chia-Hui Chen, Bei‐Chia Guo, Chiao-Po Hsu
|
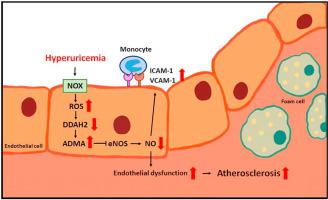
Hyperuricemia is closely associated with the mobility and mortality of patients with cardiovascular diseases. However, how hyperuricemia accelerates atherosclerosis progression is not well understood. The balance between asymmetric dimethylarginine (ADMA) and dimethylarginine dimethylaminotransferases (DDAHs) is crucial to regulate vascular homeostasis. Therefore, we investigated the role of the ADMA/DDAH pathway in hyperuricemia-induced endothelial dysfunction and atherosclerosis and the underlying molecular mechanisms in endothelial cells (ECs) and apolipoprotein E–knockout (apoe−/−) mice. Our results demonstrated that uric acid at pathological concentrations increased the intracellular levels of ADMA and downregulated DDAH-2 expression without affecting DDAH-1 expression. Excess uric acid also reduced NO bioavailability and increased monocyte adhesion to ECs, which were abolished by using the antioxidant N-acetylcysteine, the nicotinamide adenine dinucleotide phosphate oxidase inhibitor apocynin, or DDAH-2 overexpression. In apoe−/− mice, treatment with oxonic acid, a uricase inhibitor, increased the circulating level of uric acid, cholesterol, and lipid peroxidation; exacerbated systemic and aortic inflammation; and worsened atherosclerosis compared with vehicle-treated apoe−/− mice. Furthermore, oxonic acid–treated apoe−/− mice exhibited elevated ADMA plasma level and downregulated aortic expression of DDAH-2 protein. Notably, DDAH-2 overexpression in the ECs of apoe−/− mice prevented hyperuricemia-induced deleterious effects from influencing ADMA production, lipid peroxidation, inflammation, and atherosclerosis. Collectively, our findings suggest that hyperuricemia disturbs the balance of the ADMA/DDAH-2 axis, results in EC dysfunction, and, consequently, accelerates atherosclerosis.
中文翻译:
高尿酸血症通过扰乱不对称二甲基精氨酸/二甲基精氨酸二甲氨基转移酶2途径诱导内皮功能障碍并加速动脉粥样硬化
高尿酸血症与心血管疾病患者的活动能力和死亡率密切相关。然而,高尿酸血症如何加速动脉粥样硬化进展尚不清楚。不对称二甲基精氨酸 (ADMA) 和二甲基精氨酸二甲氨基转移酶 (DDAH) 之间的平衡对于调节血管稳态至关重要。因此,我们在内皮细胞 (EC) 和载脂蛋白 E 敲除 ( apoe −/− ) 小鼠中研究了 ADMA/DDAH 通路在高尿酸血症诱导的内皮功能障碍和动脉粥样硬化中的作用及其潜在分子机制。我们的结果表明,病理浓度的尿酸增加细胞内 ADMA 水平并下调 DDAH-2 表达,而不影响 DDAH-1 表达。过量的尿酸还会降低 NO 的生物利用度并增加单核细胞对 EC 的粘附,而使用抗氧化剂 N-乙酰半胱氨酸、烟酰胺腺嘌呤二核苷酸磷酸氧化酶抑制剂夹竹桃麻素或 DDAH-2 过表达可消除这些现象。在apoe −/−小鼠中,用尿酸酶抑制剂氧酸治疗会增加尿酸、胆固醇和脂质过氧化的循环水平;加剧全身和主动脉炎症;与媒介物治疗的apoe -/−小鼠相比,动脉粥样硬化恶化。此外,用氧酸处理的apoe -/−小鼠表现出 ADMA 血浆水平升高,主动脉 DDAH-2 蛋白表达下调。值得注意的是,DDAH-2 在apoe −/−小鼠EC 中的过度表达可防止高尿酸血症引起的有害影响,从而影响 ADMA 产生、脂质过氧化、炎症和动脉粥样硬化。总的来说,我们的研究结果表明,高尿酸血症会扰乱 ADMA/DDAH-2 轴的平衡,导致 EC 功能障碍,从而加速动脉粥样硬化。





























 京公网安备 11010802027423号
京公网安备 11010802027423号