当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Investigation of the Adsorption Behavior of Organic Sulfur in Coal via Density Functional Theory (DFT) Calculation and Molecular Simulation
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2021-08-18 , DOI: 10.1021/acs.jpca.1c02299 Yu Zhang 1 , Yifan Wu 1 , Jiankun Zhuo 1, 2 , Qiang Yao 1, 3
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2021-08-18 , DOI: 10.1021/acs.jpca.1c02299 Yu Zhang 1 , Yifan Wu 1 , Jiankun Zhuo 1, 2 , Qiang Yao 1, 3
Affiliation
|
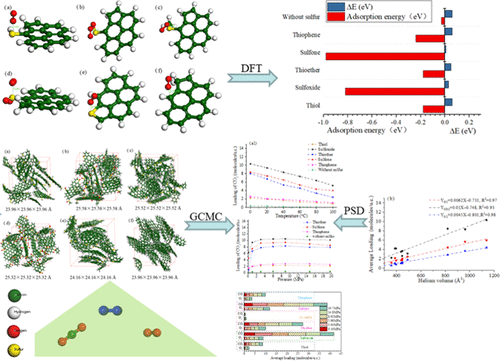
In this paper, we have investigated the chemical adsorption behavior of O2 on five types of organic sulfur (thiol, sulfoxide, thioether, sulfone, and thiophene) in polycyclic aromatic hydrocarbon (PAH) sheets using density functional theory (DFT) calculations. Here, the adsorption energy of O2-organic sulfur exceeds that of O2-PAH. Sulfone tends to be more favorable for oxidation reactions than other organic sulfur compounds and PAH by energy gap and deformation charge density analyses. A large charge transfer occurs between O2 and organic sulfur compounds by charge analysis. A radical distribution function (RDF) analysis shows that O2/CO2/N2 is preferentially adsorbed on nitrogen/sulfur/oxygen-containing functional groups in coal. To inhibit the reaction of sulfur-containing coal with oxygen, the physical adsorption of pure gas (CO2/O2/N2) and binary mixed gases (CO2 + O2/N2 + O2/CO2 + N2) is conducted at different temperatures and geological depths using molecular dynamics (MD) and grand canonical Monte Carlo (GCMC) simulations. The adsorption capacities of five types of organic sulfur with respect to the pure gases decrease with increasing temperature and increase with increasing depth. For O2/CO2, CO2/N2, and O2/N2 binary gas systems, the order with respect to adsorption amount is CO2 > O2 > N2. The factor of adsorption capacities is also evaluated, and the results show that pore volume plays a key role in adsorption behavior.
中文翻译:

通过密度泛函理论 (DFT) 计算和分子模拟研究煤中有机硫的吸附行为
在本文中,我们使用密度泛函理论 (DFT) 计算研究了 O 2在多环芳烃 (PAH) 片材中的五种有机硫(硫醇、亚砜、硫醚、砜和噻吩)上的化学吸附行为。这里,O 2 -有机硫的吸附能超过O 2 -PAH的吸附能。通过能隙和变形电荷密度分析,砜往往比其他有机硫化合物和 PAH 更有利于氧化反应。通过电荷分析,在O 2和有机硫化合物之间发生大的电荷转移。自由基分布函数 (RDF) 分析表明 O 2 /CO 2 /N 2优先吸附在煤中含氮/硫/氧的官能团上。抑制含硫煤与氧气反应,物理吸附纯气体(CO 2 /O 2 /N 2)和二元混合气体(CO 2 + O 2 /N 2 + O 2 /CO 2 + N 2 ) 是使用分子动力学 (MD) 和大正则蒙特卡罗 (GCMC) 模拟在不同温度和地质深度下进行的。五种有机硫对纯气体的吸附能力随着温度的升高而降低,随着深度的增加而增加。对于 O 2 /CO 2、CO 2 /N 2和O 2 /N 2二元气体系统,吸附量大小顺序为CO 2 > O 2 > N 2。还评估了吸附容量的因素,结果表明孔体积在吸附行为中起关键作用。
更新日期:2021-09-02
中文翻译:
通过密度泛函理论 (DFT) 计算和分子模拟研究煤中有机硫的吸附行为
在本文中,我们使用密度泛函理论 (DFT) 计算研究了 O 2在多环芳烃 (PAH) 片材中的五种有机硫(硫醇、亚砜、硫醚、砜和噻吩)上的化学吸附行为。这里,O 2 -有机硫的吸附能超过O 2 -PAH的吸附能。通过能隙和变形电荷密度分析,砜往往比其他有机硫化合物和 PAH 更有利于氧化反应。通过电荷分析,在O 2和有机硫化合物之间发生大的电荷转移。自由基分布函数 (RDF) 分析表明 O 2 /CO 2 /N 2优先吸附在煤中含氮/硫/氧的官能团上。抑制含硫煤与氧气反应,物理吸附纯气体(CO 2 /O 2 /N 2)和二元混合气体(CO 2 + O 2 /N 2 + O 2 /CO 2 + N 2 ) 是使用分子动力学 (MD) 和大正则蒙特卡罗 (GCMC) 模拟在不同温度和地质深度下进行的。五种有机硫对纯气体的吸附能力随着温度的升高而降低,随着深度的增加而增加。对于 O 2 /CO 2、CO 2 /N 2和O 2 /N 2二元气体系统,吸附量大小顺序为CO 2 > O 2 > N 2。还评估了吸附容量的因素,结果表明孔体积在吸附行为中起关键作用。





























 京公网安备 11010802027423号
京公网安备 11010802027423号