Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2021-07-29 , DOI: 10.1016/j.bioorg.2021.105200 Min Zou 1 , Jiawen Li 1 , Bo Jin 2 , Mingsheng Wang 1 , Huiping Chen 1 , Zhuangli Zhang 1 , Changzheng Zhang 1 , Zhihong Zhao 1 , Liyun Zheng 1
|
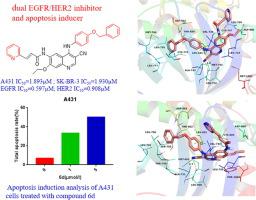
Dual targeting of EGFR/HER2 receptor is an attractive strategy for cancer therapy. Four series of 4-anilinoquinoline-3-carbonitrile derivatives were designed and prepared by introducing various functional groups, including a polar hydrophilic group (carboxylic acid), a heterocyclic substituent possessing polarity to some extent, and an unpolar hydrophobic phenyl portion, at the C-6 position of the quinoline skeleton. All of the prepared derivatives were screened for their inhibitory activities against EGFR /HER2 receptors and their antiproliferative activities against the SK-BR-3 and A431 cell lines. Compounds 6a, 6g and 6d exhibited significant activities against the target cell lines. In particular, the antiproliferative activity of 6d (IC50 =1.930 μM) against SK-BR-3 was over 2-fold higher than that of neratinib (IC50=3.966 μM), and comparable to that of Lapatinib (IC50=2.737 μM). On the other hand, 6d (IC50=1.893 μM) was more active than the reference drug Neratinib (IC50=2.151 μM), and showed comparable potency to Lapatinib (IC50=1.285μM) against A431. Cell cycle analysis and apoptosis assays indicated that 6d arrests the cell cycle in the S phase, and it is a potent apoptotic inducer. Moreover, molecular docking exhibited the binding modes of compound 6d in EGFR and HER2 binding sites, respectively. Compound 6d can be considered as a candidate for further investigation as a more potent anticancer agent.
中文翻译:
新型 4-anilinoquinoline-3-carbonitrile 衍生物作为 EGFR/HER2 双重抑制剂和凋亡诱导剂的设计、合成和抗癌评估
EGFR/HER2 受体的双重靶向是一种有吸引力的癌症治疗策略。通过在C端引入极性亲水基团(羧酸)、具有一定极性的杂环取代基和非极性疏水苯基部分,设计并制备了四个系列的4-苯胺喹啉-3-腈衍生物。 -6 喹啉骨架的位置。筛选所有制备的衍生物对EGFR/HER2受体的抑制活性和对SK-BR-3和A431细胞系的抗增殖活性。化合物6a、6g和6d对靶细胞系表现出显着的活性。尤其是6d (IC 50=1.930 μM) 比来那替尼 (IC 50 =3.966 μM)高 2 倍以上,与拉帕替尼 (IC 50 =2.737 μM)相当。在另一方面,图6d(IC 50 = 1.893μM)比参考药物来那替尼(IC多种活性50 = 2.151μM),并显示出相当的效力拉帕替尼(IC 50 =1.285μM)对A431。细胞周期分析和细胞凋亡试验表明,6d 将细胞周期阻滞在 S 期,它是一种有效的凋亡诱导剂。此外,分子对接分别在 EGFR 和 HER2 结合位点表现出化合物6d的结合模式。化合物6d 可以考虑作为更有效的抗癌剂进行进一步研究。






























 京公网安备 11010802027423号
京公网安备 11010802027423号