当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine Learning of Quasiparticle Energies in Molecules and Clusters
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-07-27 , DOI: 10.1021/acs.jctc.1c00520
Onur Çaylak 1, 2 , Björn Baumeier 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-07-27 , DOI: 10.1021/acs.jctc.1c00520
Onur Çaylak 1, 2 , Björn Baumeier 1, 2
Affiliation
|
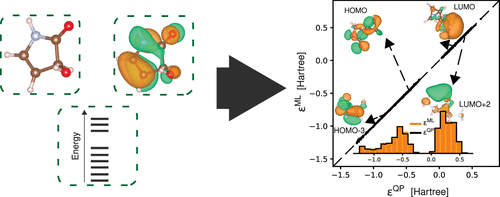
We present a Δ-machine learning approach for the prediction of GW quasiparticle energies (ΔMLQP) and photoelectron spectra of molecules and clusters, using orbital-sensitive representations (OSRs) based on molecular Cartesian coordinates in kernel ridge regression-based supervised learning. Coulomb matrix, bag-of-bond, and bond-angle-torsion representations are made orbital-sensitive by augmenting them with atom-centered orbital charges and Kohn–Sham orbital energies, both of which are readily available from baseline calculations at the level of density functional theory (DFT). We first illustrate the effects of different constructions of the OSRs on the prediction of frontier orbital energies of 22k molecules of the QM8 data set and show that it is possible to predict the full photoelectron spectrum of molecules within the data set using a single model with a mean absolute error below 0.1 eV. We further demonstrate that the OSR-based ΔMLQP captures the effects of intra- and intermolecular conformations in application to water monomers and dimers. Finally, we show that the approach can be embedded in multiscale simulation workflows, by studying the solvatochromic shifts of quasiparticle and electron–hole excitation energies of solvated acetone in a setup combining molecular dynamics, DFT, the GW approximation, and the Bethe–Salpeter equation. Our findings suggest that the ΔMLQP model allows us to predict quasiparticle energies and photoelectron spectra of molecules and clusters with GW accuracy at DFT cost.
中文翻译:

分子和团簇中准粒子能量的机器学习
我们提出了一种 Δ 机器学习方法,用于预测 GW 准粒子能量 (ΔMLQP) 以及分子和团簇的光电子能谱,在基于核岭回归的监督学习中使用基于分子笛卡尔坐标的轨道敏感表示 (OSR)。库仑矩阵、键袋和键角扭转表示通过用原子中心轨道电荷和 Kohn-Sham 轨道能量进行增强而变得对轨道敏感,这两者都可以从以下级别的基线计算中轻松获得密度泛函理论(DFT)。我们首先说明了 OSR 的不同结构对 QM8 数据集的 22k 个分子的前沿轨道能量预测的影响,并表明可以使用具有平均绝对误差低于 0.1 eV。我们进一步证明,基于 OSR 的 ΔMLQP 在应用于水单体和二聚体时捕获了分子内和分子间构象的影响。最后,我们通过在结合分子动力学、DFT、GW 近似和 Bethe-Salpeter 方程的设置中研究溶剂化丙酮的准粒子和电子空穴激发能的溶剂化变色位移,表明该方法可以嵌入到多尺度模拟工作流程中。我们的研究结果表明,ΔMLQP 模型使我们能够以 DFT 成本以 GW 精度预测分子和团簇的准粒子能量和光电子能谱。
更新日期:2021-08-10
中文翻译:
分子和团簇中准粒子能量的机器学习
我们提出了一种 Δ 机器学习方法,用于预测 GW 准粒子能量 (ΔMLQP) 以及分子和团簇的光电子能谱,在基于核岭回归的监督学习中使用基于分子笛卡尔坐标的轨道敏感表示 (OSR)。库仑矩阵、键袋和键角扭转表示通过用原子中心轨道电荷和 Kohn-Sham 轨道能量进行增强而变得对轨道敏感,这两者都可以从以下级别的基线计算中轻松获得密度泛函理论(DFT)。我们首先说明了 OSR 的不同结构对 QM8 数据集的 22k 个分子的前沿轨道能量预测的影响,并表明可以使用具有平均绝对误差低于 0.1 eV。我们进一步证明,基于 OSR 的 ΔMLQP 在应用于水单体和二聚体时捕获了分子内和分子间构象的影响。最后,我们通过在结合分子动力学、DFT、GW 近似和 Bethe-Salpeter 方程的设置中研究溶剂化丙酮的准粒子和电子空穴激发能的溶剂化变色位移,表明该方法可以嵌入到多尺度模拟工作流程中。我们的研究结果表明,ΔMLQP 模型使我们能够以 DFT 成本以 GW 精度预测分子和团簇的准粒子能量和光电子能谱。






























 京公网安备 11010802027423号
京公网安备 11010802027423号