Journal of Molecular Structure ( IF 4.0 ) Pub Date : 2021-07-20 , DOI: 10.1016/j.molstruc.2021.131091 Magaly Girão Albuquerque 1 , Raoni Schroeder B. Gonçalves 1 , Camilo Henrique da Silva Lima 1 , Fernanda Lima de Azevedo Maia 1 , Sérgio de Paula Machado 1 , Laudicéa do Nascimento Oliveira 1 , Talis Uelisson da Silva 1 , James L. Wardell 2, 3 , Solange M.S.V. Wardell 4
|
A combined structural and computational study has been conducted on two chalcone compounds, namely (E)-3-(4-bromophenyl)-1-(4,8-dimethoxynaphthalen-1-yl)prop-2-en-1-one (1), and (E)-3-(3,4-dichlorophenyl)-1-(4,8-dimethoxynaphthalen-1-yl)prop-2-en-1-one (2). These compounds are members of a series of compounds identified from a molecular modelling study as potentially active Mycobacterium tuberculosis Enoyl ACP reductase (InhA) inhibitors. In addition, the computational study indicated that the compounds have potential as optical materials.
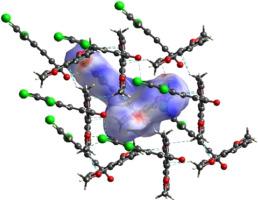
The combined study revealed that both molecules in the solid state displayed similar overall molecular conformations between a ‘‘T’’ and a ‘‘Y’’ shape, but with different arrangements in the bridging prop-2-en-1-one unit: an anti-periplanar in 1 and a syn-periplanar in 2. The experimental and calculated bond lengths and angles for anti-periplanar 1 and syn-periplanar 2 were generally in good agreement.Theoretical calculations at the B3LYP/DGTZVP level in the gas phase, DMSO and water media indicated that for both compounds, the syn-periplanar form was the energy local minimum while the anti-periplanar form was the energy global minimum. However, the energy differences are very small, with slight variations when using the different basis sets (0.80 and 0.84 kcal/mol using the B3LYP/DGTVP theory level in gas phase, 0.28 and 0.32 in DMSO and 0.27 and 0.31 kcal/mol in water). The differences in the solid state arrangements within the bridging prop-2-en-1-one unit in 1 and 2 arise from variations in the intermolecular interactions, with halogen bonds being considered to be an important factor in stabilizing a syn-periplanar arrangement in compound 2. PLATON and Hirshfeld surface analyses confirmed the important intermolecular interactions in 1 and 2.
Calculations were also carried out on the electronic and vibrational spectra, dipole moments and charge distributions of 1 and 2. The HOMO was located in the dimethoxynaphthalenyl region of the molecule with mild donor character, while the LUMO was located in the propenyl and phenyl regions of each molecule. The energy gaps between these orbitals are 3.35 eV for 1 3.25 eV for 2. The delocalization present in compounds 1 and 2 facilitates the HOMO-LUMO charge transfer process.
The total electronic molecular dipolar moments (μTotal) were calculated to be 4.1511 and 5.9316 D for 1 and 2, respectively. The values of μTotal found for 1 and 2 are both larger than reported values for similar compounds, indicating that the compounds studied here should exhibit a good non-linear optical response.
中文翻译:
两种 (E)-3-(芳基)-1-(naphthalen-1-yl)prop-2-en-1-one 查尔酮衍生物、潜在结核分枝杆菌烯酰 ACP 还原酶 (InhA) 的晶体结构、DFT 计算和 Hirshfeld 表面分析) 抑制剂和光学材料:prop-2-en-1-one 单元内的构象差异
对两种查耳酮化合物进行了结构和计算的综合研究,即 ( E )-3-(4-溴苯基)-1-(4,8-二甲氧基萘-1-基)prop-2-en-1-one ( 1 )和( E )-3-(3,4-二氯苯基)-1-(4,8-二甲氧基萘-1-基)丙-2-烯-1-酮( 2 )。这些化合物是从分子模型研究中鉴定为具有潜在活性的结核分枝杆菌烯酰 ACP 还原酶 (InhA) 抑制剂的一系列化合物的成员。此外,计算研究表明这些化合物具有作为光学材料的潜力。
联合研究表明,固态中的两种分子在“T”和“Y”形状之间显示出相似的整体分子构象,但在桥接 prop-2-en-1-one 单元中具有不同的排列:一个抗periplanar在1和SYN-periplanar在2。实验和计算的键长和角度为抗periplanar 1和SYN-periplanar 2均普遍良好agreement.Theoretical计算在气相中的B3LYP / DGTZVP水平,DMSO和水介质表明,对于两种化合物,所述顺周向形式是能量局部最小值,而反周向形式是能量局部最小值形式是能量全球最小值。然而,能量差异非常小,使用不同的基组时略有变化(使用 B3LYP/DGTVP 理论水平在气相中为 0.80 和 0.84 kcal/mol,在 DMSO 中为 0.28 和 0.32,在水中为 0.27 和 0.31 kcal/mol )。1和2 中桥接 prop-2-en-1-one 单元内固态排列的差异源于分子间相互作用的变化,卤素键被认为是稳定同周平面排列的重要因素化合物2。PLATON 和 Hirshfeld 表面分析证实了1和2 中重要的分子间相互作用。
还对1和2的电子和振动光谱、偶极矩和电荷分布进行了计算。HOMO位于分子的二甲氧基萘基区域,具有温和的供体特征,而LUMO位于每个分子的丙烯基和苯基区域。这些轨道之间的能隙为 3.35 eV 为1 3.25 eV 为2。化合物1和2 中存在的离域化促进了 HOMO-LUMO 电荷转移过程。
总电子的分子偶极矩(μ总计)分别计算为4.1511和5.9316 d为1和2,分别。μ的值总发现1和2是两个比类似的化合物报道的值越大,表明这里所研究的化合物应该表现出良好的非线性光学响应。






























 京公网安备 11010802027423号
京公网安备 11010802027423号