当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
用于计算平均力和标准结合自由能潜力的机器学习和增强采样模拟
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-07-14 , DOI: 10.1021/acs.jctc.1c00177 Martina Bertazzo 1, 2 , Dorothea Gobbo 1 , Sergio Decherchi 1, 3 , Andrea Cavalli 1, 2
Affiliation
|
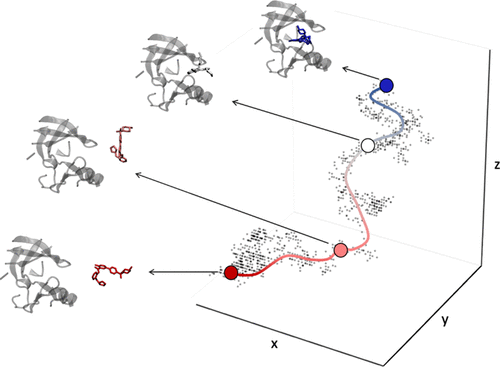
计算能力正在迅速增强,这主要是因为基于 GPU 的架构的可用性。这为热力学可观测值(包括药物与靶标结合的自由能)的系统性和鲁棒性计算创造了前所未有的模拟可能性。与目前广泛用于药物发现的相对结合自由能的计算相比,我们在这里突破了计算结合自由能和平均力潜力的界限。我们引入了一种新颖的协议,它利用增强的采样、机器学习和临时算法来限制自由能计算中的人为干预、计算时间和自由参数。我们首先在主客体系统上验证该方法,然后将该方案应用于糖原合酶激酶 3 beta(一种具有药理学意义的蛋白激酶)。总体而言,我们在相对和绝对方面与实验值具有良好的相关性。虽然我们专注于蛋白质-配体结合,但该策略广泛适用于可以用路径集体变量描述的任何复杂事件。我们系统地讨论影响最终结果的关键细节。 PLUMED-NEST 提供参数和模拟设置,以实现完全的可重复性。
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号