当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hydrodechlorination of 1,2-Dichloroethane on Platinum Catalysts: Insights from Reaction Kinetics Experiments, Density Functional Theory, and Microkinetic Modeling
ACS Catalysis ( IF 11.3 ) Pub Date : 2021-06-15 , DOI: 10.1021/acscatal.1c00940 Lang Xu 1 , Eric E. Stangland 2 , James A. Dumesic 1 , Manos Mavrikakis 1
ACS Catalysis ( IF 11.3 ) Pub Date : 2021-06-15 , DOI: 10.1021/acscatal.1c00940 Lang Xu 1 , Eric E. Stangland 2 , James A. Dumesic 1 , Manos Mavrikakis 1
Affiliation
|
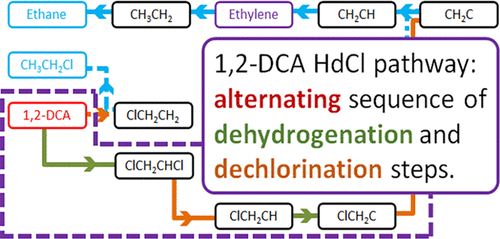
Catalytic hydrodechlorination is a promising strategy for treating industrial 1,2-dichloroethane wastes, for which Pt and Pt-based alloy catalysts are widely used. Here, we performed a detailed mechanistic study for 1,2-dichloroethane hydrodechlorination on Pt using a synergistic approach combining density functional theory (DFT) calculations, reaction kinetics experiments, and microkinetic modeling. Using planewave DFT calculations, we evaluated the reaction energy and activation energy barrier of each elementary step involved in the reaction network on Pt(111). The calculated energetics were then incorporated into a comprehensive mean-field microkinetic model accounting for a total of 65 elementary steps. The model-predicted reaction rates were compared with the results from our reaction kinetics experiments on SiO2-supported Pt catalysts. Our results indicated that the hydrodechlorination of 1,2-dichloroethane on Pt(111) starts with a H-removal step; then, it proceeds through a sequence of alternating dechlorination and dehydrogenation steps until vinylidene (CH2C*) is formed; finally, CH2C* is hydrogenated to the final product, ethane, sequentially via vinyl (CH2CH*), ethylene, and ethyl (CH3CH2*) intermediates. After model parameter adjustments, we achieved good agreement between our theoretical model and experimental results; the adjustments to the calculated parameters are consistent with the typically anticipated coverage effects. Our study offers valuable mechanistic insights, which are useful for improving catalysts for this chemistry.
中文翻译:

铂催化剂上 1,2-二氯乙烷的加氢脱氯:反应动力学实验、密度泛函理论和微动力学建模的见解
催化加氢脱氯是处理工业 1,2-二氯乙烷废物的一种有前景的策略,铂和铂基合金催化剂被广泛用于其中。在这里,我们使用结合密度泛函理论 (DFT) 计算、反应动力学实验和微动力学模型的协同方法对 Pt 上的 1,2-二氯乙烷加氢脱氯进行了详细的机理研究。使用平面波 DFT 计算,我们评估了 Pt(111) 上反应网络中涉及的每个基本步骤的反应能和活化能势垒。然后将计算出的能量学纳入一个综合平均场微动力学模型,共包含 65 个基本步骤。将模型预测的反应速率与我们在 SiO 2上的反应动力学实验结果进行比较- 负载的 Pt 催化剂。我们的结果表明 1,2-二氯乙烷在 Pt(111) 上的加氢脱氯反应是从脱氢步骤开始的。然后,它进行一系列交替的脱氯和脱氢步骤,直到形成亚乙烯基(CH 2 C*);最后,CH 2 C* 依次通过乙烯基 (CH 2 CH*)、乙烯和乙基 (CH 3 CH 2*) 中间体。经过模型参数调整,我们的理论模型与实验结果吻合较好;对计算参数的调整与通常预期的覆盖效果一致。我们的研究提供了宝贵的机械见解,有助于改进这种化学的催化剂。
更新日期:2021-07-02
中文翻译:
铂催化剂上 1,2-二氯乙烷的加氢脱氯:反应动力学实验、密度泛函理论和微动力学建模的见解
催化加氢脱氯是处理工业 1,2-二氯乙烷废物的一种有前景的策略,铂和铂基合金催化剂被广泛用于其中。在这里,我们使用结合密度泛函理论 (DFT) 计算、反应动力学实验和微动力学模型的协同方法对 Pt 上的 1,2-二氯乙烷加氢脱氯进行了详细的机理研究。使用平面波 DFT 计算,我们评估了 Pt(111) 上反应网络中涉及的每个基本步骤的反应能和活化能势垒。然后将计算出的能量学纳入一个综合平均场微动力学模型,共包含 65 个基本步骤。将模型预测的反应速率与我们在 SiO 2上的反应动力学实验结果进行比较- 负载的 Pt 催化剂。我们的结果表明 1,2-二氯乙烷在 Pt(111) 上的加氢脱氯反应是从脱氢步骤开始的。然后,它进行一系列交替的脱氯和脱氢步骤,直到形成亚乙烯基(CH 2 C*);最后,CH 2 C* 依次通过乙烯基 (CH 2 CH*)、乙烯和乙基 (CH 3 CH 2*) 中间体。经过模型参数调整,我们的理论模型与实验结果吻合较好;对计算参数的调整与通常预期的覆盖效果一致。我们的研究提供了宝贵的机械见解,有助于改进这种化学的催化剂。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号