当前位置:
X-MOL 学术
›
J. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Deciphering Reactivity and Selectivity Patterns in Aliphatic C–H Bond Oxygenation of Cyclopentane and Cyclohexane Derivatives
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2021-06-11 , DOI: 10.1021/acs.joc.1c00902 Teo Martin 1 , Marco Galeotti 1 , Michela Salamone 1 , Fengjiao Liu 2, 3 , Yanmin Yu 3, 4 , Meng Duan 3 , K N Houk 3 , Massimo Bietti 1
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2021-06-11 , DOI: 10.1021/acs.joc.1c00902 Teo Martin 1 , Marco Galeotti 1 , Michela Salamone 1 , Fengjiao Liu 2, 3 , Yanmin Yu 3, 4 , Meng Duan 3 , K N Houk 3 , Massimo Bietti 1
Affiliation
|
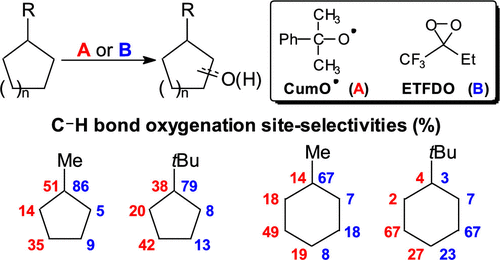
A kinetic, product, and computational study on the reactions of the cumyloxyl radical with monosubstituted cyclopentanes and cyclohexanes has been carried out. HAT rates, site-selectivities for C–H bond oxidation, and DFT computations provide quantitative information and theoretical models to explain the observed patterns. Cyclopentanes functionalize predominantly at C-1, and tertiary C–H bond activation barriers decrease on going from methyl- and tert-butylcyclopentane to phenylcyclopentane, in line with the computed C–H BDEs. With cyclohexanes, the relative importance of HAT from C-1 decreases on going from methyl- and phenylcyclohexane to ethyl-, isopropyl-, and tert-butylcyclohexane. Deactivation is also observed at C-2 with site-selectivity that progressively shifts to C-3 and C-4 with increasing substituent steric bulk. The site-selectivities observed in the corresponding oxidations promoted by ethyl(trifluoromethyl)dioxirane support this mechanistic picture. Comparison of these results with those obtained previously for C–H bond azidation and functionalizations promoted by the PINO radical of phenyl and tert-butylcyclohexane, together with new calculations, provides a mechanistic framework for understanding C–H bond functionalization of cycloalkanes. The nature of the HAT reagent, C–H bond strengths, and torsional effects are important determinants of site-selectivity, with the latter effects that play a major role in the reactions of oxygen-centered HAT reagents with monosubstituted cyclohexanes.
中文翻译:

解读环戊烷和环己烷衍生物脂肪族 C-H 键氧化的反应性和选择性模式
已经进行了枯草氧基自由基与单取代环戊烷和环己烷反应的动力学、产物和计算研究。HAT 速率、C-H 键氧化的位点选择性和 DFT 计算提供了定量信息和理论模型来解释观察到的模式。环戊烷主要在 C-1 处官能化,从甲基和叔丁基环戊烷到苯基环戊烷,叔 C-H 键活化能垒降低,与计算的 C-H BDE 一致。对于环己烷,从 C-1 开始的 HAT 的相对重要性随着从甲基-和苯基环己烷到乙基-、异丙基-和叔-丁基环己烷。在 C-2 处也观察到失活,其位点选择性随着取代基空间体积的增加逐渐转移到 C-3 和 C-4。在乙基(三氟甲基)二环氧乙烷促进的相应氧化中观察到的位点选择性支持这种机制图。将这些结果与先前获得的由苯基和叔的 PINO 自由基促进的 C-H 键叠氮化和官能化的结果进行比较-丁基环己烷与新的计算一起,为理解环烷烃的 C-H 键官能化提供了一个机制框架。HAT 试剂的性质、C-H 键强度和扭转效应是位点选择性的重要决定因素,后者的效应在以氧为中心的 HAT 试剂与单取代环己烷的反应中起主要作用。
更新日期:2021-08-07
中文翻译:
解读环戊烷和环己烷衍生物脂肪族 C-H 键氧化的反应性和选择性模式
已经进行了枯草氧基自由基与单取代环戊烷和环己烷反应的动力学、产物和计算研究。HAT 速率、C-H 键氧化的位点选择性和 DFT 计算提供了定量信息和理论模型来解释观察到的模式。环戊烷主要在 C-1 处官能化,从甲基和叔丁基环戊烷到苯基环戊烷,叔 C-H 键活化能垒降低,与计算的 C-H BDE 一致。对于环己烷,从 C-1 开始的 HAT 的相对重要性随着从甲基-和苯基环己烷到乙基-、异丙基-和叔-丁基环己烷。在 C-2 处也观察到失活,其位点选择性随着取代基空间体积的增加逐渐转移到 C-3 和 C-4。在乙基(三氟甲基)二环氧乙烷促进的相应氧化中观察到的位点选择性支持这种机制图。将这些结果与先前获得的由苯基和叔的 PINO 自由基促进的 C-H 键叠氮化和官能化的结果进行比较-丁基环己烷与新的计算一起,为理解环烷烃的 C-H 键官能化提供了一个机制框架。HAT 试剂的性质、C-H 键强度和扭转效应是位点选择性的重要决定因素,后者的效应在以氧为中心的 HAT 试剂与单取代环己烷的反应中起主要作用。
































 京公网安备 11010802027423号
京公网安备 11010802027423号