当前位置:
X-MOL 学术
›
Acta Cryst. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structural insight from intermolecular interactions and energy framework analyses of 2-substituted 6,7,8,9-tetrahydro-11H-pyrido[2,1-b]quinazolin-11-ones
Acta Crystallographica Section B ( IF 1.3 ) Pub Date : 2021-05-27 , DOI: 10.1107/s2052520621003498 Akmaljon G. Tojiboev , Burkhon Zh. Elmuradov , Halima Mouhib , Kambarali K. Turgunov , Askar Sh. Abdurazakov , Charos E. Makhmadiyarova , Bakhodir Tashkhodjaev , Sirojiddin Z. Mirzaev
Acta Crystallographica Section B ( IF 1.3 ) Pub Date : 2021-05-27 , DOI: 10.1107/s2052520621003498 Akmaljon G. Tojiboev , Burkhon Zh. Elmuradov , Halima Mouhib , Kambarali K. Turgunov , Askar Sh. Abdurazakov , Charos E. Makhmadiyarova , Bakhodir Tashkhodjaev , Sirojiddin Z. Mirzaev
|
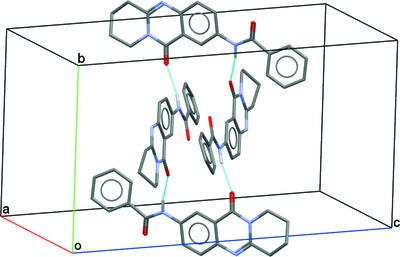
The crystal structures of three mackinazolinone derivatives (2-amino-6,7,8,9-tetrahydro-11H-pyrido[2,1-b]quinazolin-11-one at room temperature, and 2-nitro-6,7,8,9-tetrahydro-11H-pyrido[2,1-b]quinazolin-11-one and N-(11-oxo-6,8,9,11-tetrahydro-7H-pyrido[2,1-b]quinazolin-2-yl)benzamide at 100 K) are explored using X-ray crystallography. To delineate the different intermolecular interactions and the respective interaction energies in the crystal architectures, energy framework analyses were carried out using the CE-B3LYP/6-31G(d,p) method implemented in the CrystalExplorer software. In the structures the different molecules are linked by C—H…O, C—H…N and N—H…O hydrogen bonds. Together with these hydrogen bonds, C—H…π and C—O…π interactions are involved in the formation of a three-dimensional crystal network. A Hirshfeld surface analysis allows the visualization of the two-dimensional fingerprint plots and the quantification of the contributions of H…H, H…C/C…H and H…O/O…H contacts throughout the different crystal structures. To obtain additional information on the intrinsic properties of our targets and to compare the experimental crystal structures with their respective conformations in the gas phase, quantum chemical calculations at the B3LYP-D3BJ/6-311++G(d,p) level of theory, including Grimme's D3 correction term and BJ damping functions, were carried out to account for intramolecular dispersion interactions. The identified energy gaps between the highest occupied and the lowest unoccupied molecular orbitals (HOMO–LUMO gap) of our targets in the gas phase and in two implicit solvents (methanol and dimethyl sulfoxide) allow us to quantify the impact of different substituents on the reactivity of mackinazolinone derivatives.
中文翻译:

2-取代 6,7,8,9-四氢-11H-吡啶并[2,1-b]喹唑啉-11-酮分子间相互作用和能量框架分析的结构洞察
3个mackinazolinone衍生物(2-氨基-6,7,8,9-四氢11的晶体结构ħ -吡啶并[2,1- b ]喹唑啉-11-酮在室温下,和2-硝基-6,7- ,8,9-四氢-11- ħ -吡啶并[2,1- b ]喹唑啉-11-酮和ñ - (11-氧代- 6,8,9,11四氢-7- ħ -吡啶并[2,1- b ]quinazolin-2-yl)benzamide at 100 K) 使用 X 射线晶体学研究。描绘在晶体结构不同的分子间相互作用和各自的相互作用能量,能量分析框架进行了使用在实施了CE-B3LYP / 6-31G(d,p)的方法CrystalExplorer软件。在结构中,不同的分子通过 C—H…O、C—H…N 和 N—H…O 氢键连接。与这些氢键一起,C—H…π 和 C—O…π 相互作用参与了三维晶体网络的形成。Hirshfeld 表面分析允许二维指纹图的可视化以及 H…H、H…C/C…H 和 H…O/O…H 接触在不同晶体结构中的贡献的量化。为了获得关于我们目标的内在特性的更多信息,并将实验晶体结构与它们在气相中的各自构象进行比较,在 B3LYP-D3BJ/6-311++G(d,p) 理论水平上进行量子化学计算,包括 Grimme 的 D3 校正项和 BJ 阻尼函数,进行了解释分子内分散相互作用。在气相和两种隐性溶剂(甲醇和二甲基亚砜)中,我们的目标的最高占据和最低未占据分子轨道(HOMO-LUMO 间隙)之间确定的能隙使我们能够量化不同取代基对反应性的影响麦金唑啉酮衍生物。
更新日期:2021-06-07
中文翻译:
2-取代 6,7,8,9-四氢-11H-吡啶并[2,1-b]喹唑啉-11-酮分子间相互作用和能量框架分析的结构洞察
3个mackinazolinone衍生物(2-氨基-6,7,8,9-四氢11的晶体结构ħ -吡啶并[2,1- b ]喹唑啉-11-酮在室温下,和2-硝基-6,7- ,8,9-四氢-11- ħ -吡啶并[2,1- b ]喹唑啉-11-酮和ñ - (11-氧代- 6,8,9,11四氢-7- ħ -吡啶并[2,1- b ]quinazolin-2-yl)benzamide at 100 K) 使用 X 射线晶体学研究。描绘在晶体结构不同的分子间相互作用和各自的相互作用能量,能量分析框架进行了使用在实施了CE-B3LYP / 6-31G(d,p)的方法CrystalExplorer软件。在结构中,不同的分子通过 C—H…O、C—H…N 和 N—H…O 氢键连接。与这些氢键一起,C—H…π 和 C—O…π 相互作用参与了三维晶体网络的形成。Hirshfeld 表面分析允许二维指纹图的可视化以及 H…H、H…C/C…H 和 H…O/O…H 接触在不同晶体结构中的贡献的量化。为了获得关于我们目标的内在特性的更多信息,并将实验晶体结构与它们在气相中的各自构象进行比较,在 B3LYP-D3BJ/6-311++G(d,p) 理论水平上进行量子化学计算,包括 Grimme 的 D3 校正项和 BJ 阻尼函数,进行了解释分子内分散相互作用。在气相和两种隐性溶剂(甲醇和二甲基亚砜)中,我们的目标的最高占据和最低未占据分子轨道(HOMO-LUMO 间隙)之间确定的能隙使我们能够量化不同取代基对反应性的影响麦金唑啉酮衍生物。





























 京公网安备 11010802027423号
京公网安备 11010802027423号