当前位置:
X-MOL 学术
›
J. Org. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Comprehensive Computational Investigation of the Barton–Kellogg Reaction for Both Alkyl and Aryl Systems
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2021-05-20 , DOI: 10.1021/acs.joc.1c00506 Jed M. Burns 1, 2 , Timothy Clark 2 , Craig M. Williams 1
The Journal of Organic Chemistry ( IF 3.3 ) Pub Date : 2021-05-20 , DOI: 10.1021/acs.joc.1c00506 Jed M. Burns 1, 2 , Timothy Clark 2 , Craig M. Williams 1
Affiliation
|
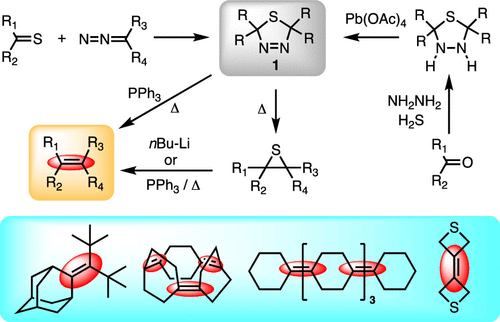
The course of the Barton–Kellogg (BK) reaction for alkyl- and aryl-substituted substrates has been investigated at the DLPNO-CCSD(T)/def2-TZVPP//ωB97X-D/def2-TZVPP level of theory, with results compared to available experimental kinetic data. Through comparison with the unsubstituted parent system, the preference for the formation of 1,3,4-dihydrothiadiazole over the isomeric 1,2,3-dihydrothiadiazole was observed to result from reduced steric repulsion in the relevant transition-state structure. Nitrogen extrusion [retro-(3 + 2)-cycloaddition] from the intermediate dihydrothiadiazole was found to be the rate-determining step. The barrier for this process was, however, significantly lower for aromatic substrates, which is consistent with the difficulty in isolating aryl-substituted dihydrothiadiazoles. The electronic structure of the transient thiocarbonyl ylide was also investigated, highlighting the contradictory results from wave-function theory- and density functional theory-based methods. Correlation of unrestricted natural orbital eigenvalues with previous experimental models suggested that the dipole intermediates possess low diradical character and are therefore considered to be closed-shell species. Exergonic conrotatory electrocyclization of the dipole led to sterically congested thiirane products, even for very bulky systems (di-t-butyl). These results complement the recent work of Mlostoń et al. Finally, DLPNO-CCSD(T)//ωB97X-D was found to be a reliable method for estimating the feasibility of the BK reaction, which should assist experimentalists in the selection of viable substrates.
中文翻译:

烷基和芳基体系巴顿-凯洛格反应的综合计算研究
已在 DLPNO-CCSD(T)/def2-TZVPP//ωB97X-D/def2-TZVPP 理论水平研究了 Barton-Kellogg (BK) 反应对烷基和芳基取代底物的反应过程,并比较了结果到可用的实验动力学数据。通过与未取代的母体系统相比,观察到 1,3,4-二氢噻二唑优于异构体 1,2,3-二氢噻二唑的形成是由于相关过渡态结构中的空间排斥降低。发现从中间体二氢噻二唑中挤出氮[逆-(3 + 2)-环加成]是决定速率的步骤。然而,该过程的障碍对于芳族底物而言明显较低,这与分离芳基取代的二氢噻二唑的困难一致。还研究了瞬态硫代羰基叶立德的电子结构,突出了基于波函数理论和密度泛函理论的方法的矛盾结果。不受限制的自然轨道特征值与以前的实验模型的相关性表明偶极子中间体具有低双自由基特性,因此被认为是闭壳物种。偶极子的放能旋转电环化导致空间拥挤的硫杂丙环产品,即使对于非常庞大的系统(双- 不受限制的自然轨道特征值与以前的实验模型的相关性表明偶极子中间体具有低双自由基特性,因此被认为是闭壳物种。偶极子的放能旋转电环化导致空间拥挤的硫杂丙环产品,即使对于非常庞大的系统(双- 不受限制的自然轨道特征值与以前的实验模型的相关性表明偶极子中间体具有低双自由基特性,因此被认为是闭壳物种。偶极子的放能旋转电环化导致空间拥挤的硫杂丙环产品,即使对于非常庞大的系统(双-叔丁基)。这些结果补充了 Mlostoń 等人最近的工作。最后,发现 DLPNO-CCSD(T)//ωB97X-D 是一种可靠的估计 BK 反应可行性的方法,应该有助于实验人员选择可行的底物。
更新日期:2021-06-04
中文翻译:
烷基和芳基体系巴顿-凯洛格反应的综合计算研究
已在 DLPNO-CCSD(T)/def2-TZVPP//ωB97X-D/def2-TZVPP 理论水平研究了 Barton-Kellogg (BK) 反应对烷基和芳基取代底物的反应过程,并比较了结果到可用的实验动力学数据。通过与未取代的母体系统相比,观察到 1,3,4-二氢噻二唑优于异构体 1,2,3-二氢噻二唑的形成是由于相关过渡态结构中的空间排斥降低。发现从中间体二氢噻二唑中挤出氮[逆-(3 + 2)-环加成]是决定速率的步骤。然而,该过程的障碍对于芳族底物而言明显较低,这与分离芳基取代的二氢噻二唑的困难一致。还研究了瞬态硫代羰基叶立德的电子结构,突出了基于波函数理论和密度泛函理论的方法的矛盾结果。不受限制的自然轨道特征值与以前的实验模型的相关性表明偶极子中间体具有低双自由基特性,因此被认为是闭壳物种。偶极子的放能旋转电环化导致空间拥挤的硫杂丙环产品,即使对于非常庞大的系统(双- 不受限制的自然轨道特征值与以前的实验模型的相关性表明偶极子中间体具有低双自由基特性,因此被认为是闭壳物种。偶极子的放能旋转电环化导致空间拥挤的硫杂丙环产品,即使对于非常庞大的系统(双- 不受限制的自然轨道特征值与以前的实验模型的相关性表明偶极子中间体具有低双自由基特性,因此被认为是闭壳物种。偶极子的放能旋转电环化导致空间拥挤的硫杂丙环产品,即使对于非常庞大的系统(双-叔丁基)。这些结果补充了 Mlostoń 等人最近的工作。最后,发现 DLPNO-CCSD(T)//ωB97X-D 是一种可靠的估计 BK 反应可行性的方法,应该有助于实验人员选择可行的底物。






























 京公网安备 11010802027423号
京公网安备 11010802027423号