当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
分子内电荷转移激发的参考能量
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-05-06 , DOI: 10.1021/acs.jctc.1c00226
Pierre-François Loos 1 , Massimiliano Comin 2 , Xavier Blase 2 , Denis Jacquemin 3
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2021-05-06 , DOI: 10.1021/acs.jctc.1c00226
Pierre-François Loos 1 , Massimiliano Comin 2 , Xavier Blase 2 , Denis Jacquemin 3
Affiliation
|
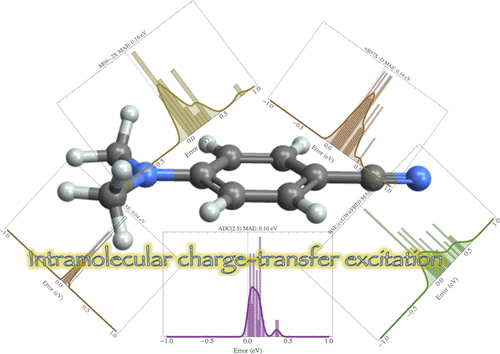
为了完成我们之前致力于有机化合物中局部跃迁和里德堡跃迁的努力,我们为(π-共轭)分子化合物中发生的分子内电荷转移跃迁提供了一系列高度准确的垂直跃迁能量。为此,我们应用了一个复合协议,该协议由由 Dunning 双 ζ 基组确定的线性响应 CCSDT 激发能量组成,该基组由 CC3/CCSDT-3 能量校正,由相应的三 ζ 基获得。在 CCSD 和 CC2 级别获得了进一步的基组校正(高达 aug-cc-pVQZ)。我们报告了在 17 种化合物(氨基苄腈、苯胺、薁、苄腈、苯并噻二唑、二甲氨基苄腈、二甲基苯胺、二肽、β-二肽、氯化氢、硝基苯胺、硝基苯、硝基二甲基苯胺、N-氧化物、N-苯基吡咯、酞嗪和喹喔啉]。然后使用这些参考值对一系列波函数进行基准测试 [CIS(D)、SOPPA、RPA(D)、EOM-MP2、CC2、CCSD、CCSD(T)(a)*、CCSDR(3)、CCSDT- 3、CC3、ADC(2)、ADC(3) 和 ADC(2.5)],基于格林函数的 Bethe-Salpeter 方程 (BSE) 形式主义在部分自洽 ev GW方案之上执行,考虑两种不同的起始积分(BSE/ev GW @HF 和 BSE/ev GW@PBE0) 和瞬态密度泛函理论 (TD-DFT) 结合几个交换相关泛函(B3LYP、PBE0、M06-2X、CAM-B3LYP、LC-ωHPBE、ωB97X、ωB97X-D 和 M11) . 结果表明,包括三元组的 CC 方法,即 CCSD(T)(a)*、CCSDR(3)、CCSDT-3 和 CC3,提供了相当小的平均偏差(≤0.10 eV),CC3 成为唯一的化学准确的方法。ADC(2.5) 也表现良好,O (N 6 ) 形式缩放的平均绝对误差为 0.11 eV ,而 CC2 和 BSE/ev GW @PBE0 也提供了非常令人满意的结果,因为它们各自的O (N 5 ) 和O ( ñ 4) 计算缩放。在 TD-DFT 上下文中,表现最好的泛函是 ωB97X-D,紧随其后的是 CAM-B3LYP 和 M06-2X,相对于理论最佳估计,它们都提供大约 0.15 eV 的平均绝对误差。

"点击查看英文标题和摘要"
更新日期:2021-06-08
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号