当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Influence of Fe and Ni Doping on the OER Performance at the Co3O4(001) Surface: Insights from DFT+U Calculations
ACS Catalysis ( IF 11.3 ) Pub Date : 2021-04-22 , DOI: 10.1021/acscatal.1c00214 Yuman Peng 1 , Hamidreza Hajiyani 1 , Rossitza Pentcheva 1
ACS Catalysis ( IF 11.3 ) Pub Date : 2021-04-22 , DOI: 10.1021/acscatal.1c00214 Yuman Peng 1 , Hamidreza Hajiyani 1 , Rossitza Pentcheva 1
Affiliation
|
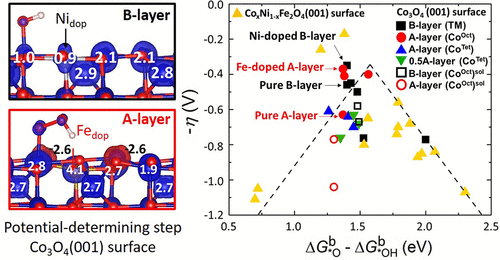
Using density functional theory calculations with an on-site Hubbard U term, we study the oxygen evolution reaction (OER) at the Co3O4(001) surface. The stability of different surface terminations as a function of oxygen partial pressure as well as pH and applied voltage is compiled in a Pourbaix diagram. The termination with octahedral Co and O ions (B-layer) is found to have the lowest overpotential of 0.46 V for an octahedral Co reaction site, associated with its p-type conducting character and the higher oxidation state of the active site (+4) during OER. Furthermore, we systematically investigated the effect of Fe and Ni doping on the overpotential. Our results indicate that Ni doping at an octahedral site in the surface layer reduces the overpotential from 0.46 to 0.34 V. Likewise, Fe doping at an octahedral site at the tetrahedral Co termination (A-layer) lowers η from 0.63 to 0.37 V with octahedral Co remaining in the active site. We note that the potential determining step changes from *OH (B-layer) to *OOH formation (A-layer). While implicit solvation increases the overpotential by 0.2 V (B-layer) and 0.4 V (A-layer), which is attributed to enhanced binding energies of the intermediates, the general trends with respect to doping remain unchanged. The scaling relationship of the binding energies of *OOH and *OH is overall satisfied, with the doped systems lying close to the top of the volcano plot of the overpotential versus (ΔG*Ob – ΔG*OHb). A further insight into the origin of this behavior is gained by analyzing the changes in oxidation states of surface ions and, in particular, the Co active site during OER.
中文翻译:

Fe和Ni掺杂对Co 3 O 4(001)表面OER性能的影响:DFT + U计算得出的见解
使用现场Hubbard U项的密度泛函理论计算,我们研究了Co 3 O 4处的析氧反应(OER)(001)表面。在Pourbaix图中汇总了不同表面终端的稳定性与氧分压以及pH和施加电压的关系。发现八面体Co和O离子的终止层(B层)对于八面体Co反应位点具有0.46 V的最低过电势,这与其p型导电特性和活性位点的较高氧化态有关(+4 )。此外,我们系统地研究了铁和镍掺杂对过电势的影响。我们的结果表明,在表面层的八面体位置处掺杂Ni会使过电势从0.46 V降低到0.34V。同样,在四面体Co端接处(A层)的八面体位置处的Fe掺杂会使η从0.63降至0.37 V(八面体) Co保留在活动站点中。我们注意到,电位确定步骤从* OH(B层)变为* OOH形成(A层)。虽然隐式溶剂化使过电势增加了0.2 V(B层)和0.4 V(A层),这归因于中间体的结合能增强,但有关掺杂的一般趋势仍未改变。* OOH和* OH的结合能的比例关系总体上得到满足,掺杂系统位于超电势与(Δ)的火山图顶部附近ģ *○ b - Δ ģ * OH b)。通过分析表面离子(特别是在OER期间)中Co活性位点的氧化态变化,可以进一步了解这种行为的起源。
更新日期:2021-05-07
中文翻译:
Fe和Ni掺杂对Co 3 O 4(001)表面OER性能的影响:DFT + U计算得出的见解
使用现场Hubbard U项的密度泛函理论计算,我们研究了Co 3 O 4处的析氧反应(OER)(001)表面。在Pourbaix图中汇总了不同表面终端的稳定性与氧分压以及pH和施加电压的关系。发现八面体Co和O离子的终止层(B层)对于八面体Co反应位点具有0.46 V的最低过电势,这与其p型导电特性和活性位点的较高氧化态有关(+4 )。此外,我们系统地研究了铁和镍掺杂对过电势的影响。我们的结果表明,在表面层的八面体位置处掺杂Ni会使过电势从0.46 V降低到0.34V。同样,在四面体Co端接处(A层)的八面体位置处的Fe掺杂会使η从0.63降至0.37 V(八面体) Co保留在活动站点中。我们注意到,电位确定步骤从* OH(B层)变为* OOH形成(A层)。虽然隐式溶剂化使过电势增加了0.2 V(B层)和0.4 V(A层),这归因于中间体的结合能增强,但有关掺杂的一般趋势仍未改变。* OOH和* OH的结合能的比例关系总体上得到满足,掺杂系统位于超电势与(Δ)的火山图顶部附近ģ *○ b - Δ ģ * OH b)。通过分析表面离子(特别是在OER期间)中Co活性位点的氧化态变化,可以进一步了解这种行为的起源。
































 京公网安备 11010802027423号
京公网安备 11010802027423号