当前位置:
X-MOL 学术
›
Energy Fuels
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
噻吩和呋喃的单分子热解机理:从头算比较
Energy & Fuels ( IF 5.2 ) Pub Date : 2021-04-20 , DOI: 10.1021/acs.energyfuels.1c00291
Tianshuang Li 1, 2 , Jie Li 1, 2 , Hongliang Zhang 1, 2 , Jingkun Wang 3 , Jin Xiao 1, 2
Energy & Fuels ( IF 5.2 ) Pub Date : 2021-04-20 , DOI: 10.1021/acs.energyfuels.1c00291
Tianshuang Li 1, 2 , Jie Li 1, 2 , Hongliang Zhang 1, 2 , Jingkun Wang 3 , Jin Xiao 1, 2
Affiliation
|
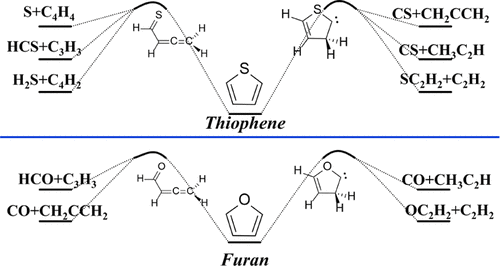
噻吩硫是石油中最稳定,含量最高的有机硫。在这项工作中,使用高水平的从头算方法研究了噻吩的单分子热解机理。相比之下,使用相同的方法计算出呋喃的相似反应。发现噻吩单分子热解最可能的引发反应是2,3-H和3,2-H转移。3,2-H移位形成buta-2,3-dienethial,随后分解为CS + CH 3 C 2 H和HC 2 CH 2自由基(*)+ HCS *,而2,3-H迁移对应于α-卡宾并最终导致C 2 H 2 + SCCH 2,CS + CH 3 C 2H,HC 2 CH 2 * + HCS *和S + C 2 CHC 2 H.ħ 2 S被假定通过副反应产生来。结合热力学,推断出优选的产物为C 2 H 2 + SCCH 2。由于反应途径的较高的能量屏障和产物的较低的热稳定性,噻吩在动力学和热力学上更稳定。

"点击查看英文标题和摘要"
更新日期:2021-05-06
"点击查看英文标题和摘要"


































 京公网安备 11010802027423号
京公网安备 11010802027423号